Nội dung toàn văn Tiêu chuẩn quốc gia TCVN 10547:2014 (ISO/TS 22367:2008) về Phòng thí nghiệm y tế - Giảm sai lỗi thông qua quản lý rủi ro và cải tiến liên tục
TIÊU CHUẨN QUỐC GIA
TCVN 10547:2014
ISO/TS 22367:2008
PHÒNG THÍ NGHIỆM Y TẾ - GIẢM SAI LỖI THÔNG QUA QUẢN LÝ RỦI RO VÀ CẢI TIẾN LIÊN TỤC
Medical laboratories — Reduction of error through risk management and continual improvement
Lời nói đầu
TCVN 10547:2014 hoàn toàn tương đương với ISO/TS 22367:2008 và đính chính kỹ thuật 1:2009.
TCVN 10547:2014 do Ban kỹ thuật Tiêu chuẩn Quốc gia TCVN/TC 176 Quản lý chất lượng và đảm bảo chất lượng biên soạn, Tổng cục Tiêu chuẩn Đo lường Chất lượng đề nghị, Bộ Khoa học và Công nghệ công bố.
Lời giới thiệu
Một yêu cầu trong TCVN ISO 15189 là phòng thí nghiệm phải có một quá trình nghiên cứu nhằm nhận biết các khía cạnh không phù hợp với quy trình của tổ chức hoặc với các yêu cầu đã định trong hệ thống quản lý chất lượng. TCVN ISO 15189 quy định rằng việc này phải được kết nối với cả hành động khắc phục và hành động phòng ngừa. Ngoài ra, tiêu chuẩn quy định rằng lãnh đạo xem xét sự phù hợp và hiệu lực của hệ thống và các hoạt động của hệ thống để hỗ trợ chăm sóc bệnh nhân cũng như đưa ra những thay đổi cần thiết. Việc này có thể thực hiện một cách tốt nhất thông qua việc xem xét đưa rủi ro tiềm ẩn vào từng bước của mỗi quá trình.
Hành động phòng ngừa được hoạch định và thích hợp với các quá trình phòng ngừa, trên cơ sở các thông tin có thể kiểm tra xác nhận và được thực hiện nhằm ngăn ngừa diễn ra hành động tiềm ẩn. Hành động khắc phục cũng được hoạch định theo cách tương tự cùng với quá trình thực hiện thích hợp; tuy nhiên, những hành động này được thực hiện để sửa chữa các vấn đề đã được nhận biết và tránh tái diễn. Quản lý rủi ro là quá trình hoạch định và là một phần của hành động phòng ngừa, khắc phục.
Hành động phòng ngừa và khắc phục có thể được định hướng hiệu lực hơn nếu chúng được dựa trên các thông tin được sắp xếp tốt; hệ thống phân loại và phân tích quản lý rủi ro là hai quá trình giúp cung cấp các thông tin được sắp xếp tốt.
Về mặt quản lý của tổ chức, rủi ro được mô tả là mối quan tâm đa chiều về tính ổn định và khả năng dự báo kết quả. Rủi ro của tổ chức bao gồm các thành phần ảnh hưởng tới các khía cạnh vận hành, kỹ thuật, trách nhiệm pháp lý của phòng thí nghiệm. Về mặt cải tiến liên tục, các yếu tố rủi ro về nguy cơ thiệt hại được xếp cao hơn các yếu tố về lợi ích thu được. Xem xét rủi ro cần đưa vào các yếu tố có sự liên kết nhưng khác nhau về khả năng xảy ra và mức độ nghiêm trọng của tác động. Nhân tố tác động ngay khi có rủi ro có thể thực hiện trực tiếp hoặc gián tiếp.
Khuôn khổ quản lý rủi ro có thể được mô tả gồm các bước sau:
a) hoạch định rủi ro;
b) nhận diện rủi ro và tác động của rủi ro;
c) xây dựng chiến lược xử lý rủi ro; và d) theo dõi việc kiểm soát rủi ro.
Những bước này nhất quán với các yêu cầu quản lý nêu trong TCVN ISO 15189, bao gồm:
- nhận biết và kiểm soát sự không phù hợp;
- thiết lập hành động khắc phục và phòng ngừa;
- thực hiện đánh giá nội bộ và xem xét của lãnh đạo; và
- thực hiện cải tiến liên tục
Tiêu chuẩn này nhằm đưa ra những bước đầu tiên để đưa quản lý rủi ro vào cơ cấu, tổ chức, hoạt động và hệ thống quản lý của phòng thí nghiệm y tế.
Việc phân loại sự không phù hợp, sai lỗi và sự cố là hữu ích cho mục đích theo dõi và cho phép phòng thí nghiệm xác định mức độ nghiêm trọng của sự không phù hợp nhằm thiết lập thứ tự ưu tiên giải quyết và nhận biết các yếu tố nguyên nhân cơ bản góp phần gây sai lỗi.
Thường áp dụng các xem xét thuộc quy định của địa phương, khu vực và quốc gia.
PHÒNG THÍ NGHIỆM Y TẾ - GIẢM SAI LỖI THÔNG QUA QUẢN LÝ RỦI RO VÀ CẢI TIẾN LIÊN TỤC
Medical laboratories — Reduction of error through risk management and continual improvement
1. Phạm vi áp dụng
Tiêu chuẩn này mô tả việc áp dụng TCVN ISO 15189 như một hệ thống làm giảm sai lỗi trong phòng thí nghiệm và nâng cao sự an toàn cho bệnh nhân bằng cách áp dụng các quy tắc quản lý rủi ro viện dẫn tới những khía cạnh xét nghiệm của chu kỳ chăm sóc y tế của phòng thí nghiệm, đặc biệt là các khía cạnh trước và và sau xét nghiệm. Tiêu chuẩn này đưa ra phương pháp phát hiện và mô tả đặc trưng lỗi của phòng thí nghiệm y tế có thể tránh được nhờ áp dụng TCVN ISO 15189.
2. Tài liệu viện dẫn
Các tài liệu viện dẫn dưới dây đều cần thiết cho việc áp dụng tiêu chuẩn này. Đối với tài liệu viện dẫn ghi năm công bố thì áp dụng bản được nêu. Đối với tài liệu viện dẫn không ghi năm công bố thì áp dụng bản mới nhất bao gồm cả các sửa đổi.
TCVN ISO 9000 (ISO 9000), Hệ thống quản lý chất lượng – Cơ sở và từ vựng
TCVN 8023:2009 (ISO 14971:2007), Trang thiết bị y tế - Áp dụng quản lý rủi ro đối với trang thiết bị y tế
TCVN ISO 15189 (ISO 15189), Phòng thí nghiệm y tế - Yêu cầu đối với chất lượng và năng lực
TCVN 9788 (ISO/IEC Guide 73), Quản lý rủi ro – Từ vựng – Hướng dẫn sử dụng
3. Thuật ngữ và định nghĩa
Tiêu chuẩn này áp dụng các thuật ngữ và định nghĩa nêu trong TCVN ISO 9000, TCVN 8023 (ISO 14971:2007), TCVN ISO 15189, TCVN 9788 (ISO/IEC Guide 73) và thuật ngữ, định nghĩa dưới đây.
3.1. Lỗi phòng thí nghiệm (laboratory error)
Sai lỗi của một hành động được hoạch định cần hoàn thành theo dự kiến hoặc việc sử dụng kế hoạch sai để đạt được mục tiêu, xảy ra ở phần bất kỳ của chu kỳ phòng thí nghiệm, từ yêu cầu xét nghiệm đến báo cáo kết quả, diễn giải hợp lý cũng như tác động trở lại chúng.
3.2. Lỗi tác nghiệp (active error) Lỗi từ người thao tác trực tiếp.
CHÚ THÍCH: Xem tài liệu tham khảo [2].
3.3. Lỗi nhận thức (cognitive error)
Lỗi từ sự lựa chọn không đúng, do thiếu kiến thức, hiểu sai thông tin sẵn có hoặc áp dụng nguyên tắc nhận thức sai.
CHÚ THÍCH 1: Xem tài liệu tham khảo [1].
CHÚ THÍCH 2: Lỗi nhận thức còn được gọi là “lỗi chủ ý” hay “lầm lẫn” (xem tài liệu tham khảo [9]).
3.4. Phân tích phương thức và tác động của sai lỗi (failure modes and effects analysis FMEA)
Việc xem xét có phương pháp một hệ thống hay sản phẩm liên quan đến xác định các sai lỗi tiềm ẩn và đánh giá ảnh hưởng của sai lỗi tới toàn bộ việc thực hiện hệ thống/sản phẩm.
CHÚ THÍCH 1: Phân tích này bao gồm một hoặc nhiều đánh giá về các bước tiến hành phòng tránh sai lỗi hoặc để giảm thiểu ảnh hưởng của nó.
CHÚ THÍCH 2: Quy trình này đôi khi được gọi là phân tích “từ dưới lên”.
3.5. Lỗi tiềm ẩn (latent error)
Lỗi do các yếu tố cấu trúc tiềm ẩn không thuộc quyền kiểm soát của người vận hành quan trọng cuối cùng.
VÍ DỤ: thiết bị không tốt, thiết kế kém, quyết định quản lý, hoặc cơ cấu tổ chức (xem tài liệu tham khảo [2]).
3.6. Lỗi không chủ ý (non-cognitive error)
Lỗi do sai sót vô ý hay vô thức trong hành vi dự kiến tự động.
CHÚ THÍCH 1: Xem tài liệu tham khảo [1].
CHÚ THÍCH 2: Lỗi không chủ ý còn được gọi là “lỗi sơ đồ” hay “trượt” (xem tài liệu tham khảo [9]).
4. Trách nhiệm của lãnh đạo đối với hành động khắc phục, phòng ngừa và cải tiến liên tục
4.1. Khái quát
Lãnh đạo cần đảm bảo cung cấp đủ nguồn lực nhằm chắc chắn rằng tất cả các hành động khắc phục và phòng ngừa đều được xác định và tiến hành.
4.2. Trách nhiệm của lãnh đạo trong hành động phòng ngừa
Lãnh đạo cần:
- xác định chính sách và các quá trình thu thập dữ liệu về việc thực hiện quá trình qua chu kỳ thử nghiệm,
- phân tích dữ liệu theo xu hướng và mẫu để đề xuất khả năng xảy ra các vấn đề hoặc lỗi, và
- xây dựng và thực hiện các hành động phòng ngừa thông qua cải tiến quá trình để loại bỏ nguyên nhân của sự không phù hợp tiềm ẩn nhằm tránh xảy ra.
4.3. Trách nhiệm của lãnh đạo trong hành động khắc phục
Lãnh đạo cần:
- xác định chính sách và các quá trình để nhận biết và báo cáo về sự không phù hơp, lỗi và sự cố,
- đảm bảo tất cả nhân viên được đào tạo để có thể nhận biết đúng và báo cáo về sự không phù hợp, lỗi và sự cố,
- xem xét kết quả phân tích sự không phù hợp, lỗi và sự cố, và
- xây dựng các hành động khắc phục và sửa chữa để loại bỏ hoặc giảm sự tái diễn sự không phù hợp, lỗi và sự cố.
4.4. Trách nhiệm của lãnh đạo trong cải tiến liên tục
Lãnh đạo cần đảm bảo để kết quả quản lý rủi ro, hành động phòng ngừa và hành động khắc phục được kết hợp vào quá trình cải tiến liên tục.
5. Nhận biết sự không phù hợp, lỗi và sự cố thực tế và tiềm ẩn của phòng thí nghiệm
5.1. Những sự không phù hợp, lỗi và sự cố thực tế và tiềm ẩn của phòng thí nghiệm cần được xác định bằng các quá trình sau đây:
- xem xét đánh giá nội bộ,
- báo cáo sự cố,
- cơ hội cải tiến, hoặc
- quá trình phân tích rủi ro tiềm ẩn.
5.2. Sơ đồ của toàn bộ quá trình phân tích có thể được sử dụng để xác định nguyên nhân thực tế và tiềm ẩn dẫn đến các kết quả sai. Từng bước của quá trình này cần được phân tích để ước lượng xác suất cho mỗi mối nguy (xem Phụ lục A).
6. Phân loại sự không phù hợp, lỗi và sự cố của phòng thí nghiệm
Có thể phân loại những sự không phù hợp, lỗi và sự cố của phòng thí nghiệm đã được xác định. Các điểm phân loại được liệt kê dưới đây, nhưng không giới hạn ở:
a) Giai đoạn chu kì của sự kiện:
- Trước xét nghiệm:
• xác định bệnh nhân không đúng;
• thông tin chẩn đoán sai hoặc thiếu sót;
• giải thích sai về y lệnh;
• chuẩn bị bệnh nhân không đúng;
• lựa chọn sai chất bảo quản hoặc dụng cụ chứa đựng;
• chọn sai nhãn bao bì;
• pha trộn mẫu không đúng;
• lựa chọn sai thời gian;
• điều kiện hay thời gian vận chuyển không đúng.
- Xét nghiệm:
• kết quả kiểm soát chất lượng khác nhau;
• không phù hợp về thủ tục;
• lỗi thiết bị hoặc thuốc thử;
• trì hoãn thời gian hoàn thành (thời gian trả kết quả).
CHÚ THÍCH: Trì hoãn thời gian có thể xảy ra trong toàn bộ chu kỳ phòng thí nghiệm.
- Sau xét nghiệm:
• kết quả không đúng;
• sao chép kết quả không đúng;
• báo cáo không rõ ràng;
• gán nhầm kết quả cho bệnh nhân;
• báo cáo gửi không đúng người;
• thiếu thông tin về những hạn chế trong giải thích kết quả.
b) Thừa nhận sự không phù hợp, lỗi hay sự cố của phòng thí nghiệm:
- nội bộ hoặc bên ngoài phòng thí nghiệm.
c) Trách nhiệm đối với sự kiện:
- lỗi tác nghiệp hoặc tiềm ẩn;
- lỗi nhận thức hoặc không chủ ý;
- nội bộ hoặc bên ngoài phòng thí nghiệm, hay không thể xác định.
d) Khả năng phòng ngừa:
- từ không phòng ngừa được đến khả năng phòng ngừa cao.
e) Ảnh hưởng đến chăm sóc bệnh nhân:
- không ảnh hưởng hay ảnh hưởng tối thiểu;
- dẫn đến điều trị hoặc chẩn đoán chậm;
- dẫn đến điều trị hoặc chẩn đoán không phù hợp.
7. Hành động phòng ngừa và hành động khắc phục
7.1. Việc xác định những sai lỗi tiềm ẩn và sự không phù hợp của phòng thí nghiệm thông qua việc xem xét theo kế hoạch các quá trình và việc xác định tác động của những thay đổi có hiệu quả cao trong ngăn ngừa xảy ra lỗi. Sự không phù hợp, lỗi và sự cố của phòng thí nghiệm cần được xác định bằng biện pháp xem xét việc đánh giá nội bộ, báo cáo sự cố, cơ hội cải tiến hoặc quá trình phân tích rủi ro tiềm năng.
7.2. Khi một sự không phù hợp của phòng thí nghiệm được xác định, hành động khắc phục cần làm giảm sự tái diễn của sự không phù hợp hay sự phát sinh sự không phù hợp liên quan, nếu giai đoạn điều tra của hành động khắc phục để bao gồm việc phân tích nguyên nhân gốc rễ góp phần gây nên sự không phù hợp.
CHÚ THÍCH: Điều này khuyến khích một phân tích kỹ lưỡng không chỉ về nguyên nhân có thể của lỗi, mà cả về những yếu tố có thể đã góp phần dẫn đến xuất hiện nguyên nhân. Điều này còn khuyến khích kế hoạch hành động khắc phục đưa ra tất cả các yếu tố đóng góp.
8. Đánh giá rủi ro nảy sinh từ sự không phù hợp thực tế và tiềm ẩn của phòng thí nghiệm
Người quản lý chất lượng cần thiết lập và duy trì các quá trình để:
a) nhận biết những quá trình có rủi ro cao ở đó sự tiềm ẩn về lỗi có thể dẫn đến nguy cơ mất an toàn cho bệnh nhân;
b) nhận biết sự cố thực tế liên quan đến sai lệch so với yêu cầu tiêu chuẩn;
c) dự đoán và đánh giá những nguy cơ liên quan đến sự an toàn của bệnh nhân;
d) kiểm soát những nguy cơ này; và
e) theo dõi hiệu lực của việc kiểm soát.
Quá trình này cần xem xét mô hình rủi ro (xem Phụ lục B) và bao gồm phương pháp để đánh giá nguy cơ sai lỗi của quá trình (xem Phụ lục C).
CHÚ THÍCH 1: Phân tích rủi ro tiềm ẩn có thể là một FMEA, hoặc những công cụ khác để xác định sự tiềm ẩn về lỗi, vấn đề hay nguy cơ an toàn đối với bệnh nhân. Ví dụ phân tích mối nguy quá trình (PHA), phân tích mức độ nghiêm trọng và ảnh hưởng của phương thức sai lỗi [FME(C)A], phân tích cây lỗi (FTA), phân tích mối nguy và khả năng hoạt động (HAZOP) và phân tích mối nguy và điểm kiểm soát tới hạn (HACCP). TCVN 8023:2008 (ISO 14971:2007), Phụ lục G, đưa ra thảo luận về những công cụ này và khả năng áp dụng của chúng. Xem thêm các tài liệu tham khảo [13], [14], [15], [16], [17] và [18].
Quá trình này cần phải bao gồm cả việc đánh giá các quá trình có khả năng rủi ro cao, trên cơ sở tiền đánh giá, khảo sát, kinh nghiệm hay tư liệu dựa trên bằng chứng về thủ tục khi sai lỗi có thể dẫn đến nguy cơ về an toàn nghiêm trọng cho bệnh nhân.
Người quản lý về chất lượng cần xác định một nhóm để nghiên cứu về quá trình được lựa chọn.
CHÚ THÍCH 2: Khuyến khích các thành viên trong nhóm có kiến thức cá nhân về quá trình và ảnh hưởng của sai lỗi.
CHÚ THÍCH 3: Khuyến khích nhóm bao gồm những người có trình độ và kiến thức phù hợp. Nhóm cần phải tổ chức một phân tích kỹ lưỡng về quá trình bao gồm:
- từng hoạt động của quá trình,
- cách mà mỗi hoạt động của quá trình có thể sai sót,
- cách mà mỗi sai sót ở từng hoạt động có thể ảnh hưởng đến sự an toàn của bệnh nhân,
- sự nghiêm trọng và khả năng xảy ra của mỗi ảnh hưởng của phương thức sai lỗi,
- những ảnh hưởng của dạng sai sót quan trọng nhất,
- nguyên nhân gốc rễ tiềm ẩn của những ảnh hưởng của dạng sai sót quan trọng nhất, và
- hành động dẫn đến nguyên nhân gốc rễ.
CHÚ THÍCH 4: Mức độ nghiêm trọng có thể được phân loại thành không đáng kể, nhỏ, quan trọng, nghiêm trọng và thảm họa. Khả năng xảy ra có thể được phân loại theo không thể xảy ra, rất nhỏ, không thường xuyên, có thể xảy ra và xảy ra thường xuyên.
Phân tích FMEA cần tạo thành cơ sở của một kế hoạch hành động phòng ngừa cho những vấn đề tiềm ẩn, hoặc kế hoạch hành động khắc phục cho những vấn đề đã xảy ra.
9. Xem xét sự không phù hợp, lỗi và sự cố phòng thí nghiệm thu thập được
Định kỳ, nội dung của kế hoạch hành động khắc phục cần được xem xét để xác định các yếu tố chung và các vấn đề tiếp theo liên quan đến sự không phù hợp, lỗi và sự cố phòng thí nghiệm.
Các yếu tố nguyên nhân cơ bản của sự không phù hợp, lỗi và sự cố phòng thí nghiệm cần được phân tích một cách phù hợp (xem Phụ lục C).
Phân tích này cần được đưa vào hành động phòng ngừa, hành động khắc phục và kế hoạch cải tiến liên tục thích hợp.
10. Kế hoạch hành động phòng ngừa và hành động khắc phục
Lãnh đạo phòng thí nghiệm cần chuẩn bị một kế hoạch điều tra và phòng ngừa/khắc phục mọi sự không phù hợp, lỗi hay sự cố được xác định trong FMEA hoặc được quan sát bằng một cách khác. Kế hoạch đó cần bao gồm:
- phạm vi của kế hoạch,
- mô tả ảnh hưởng của phương thức sai lỗi chi tiết, sự không phù hợp, lỗi hay sự cố,cụ thể,
- việc nhận biết những rủi ro tiềm năng liên quan đến lỗi hoặc sự không phù hợp tiềm năng,
- phân công trách nhiệm để thay đổi theo yêu cầu,
- yêu cầu đối với việc xem xét,
- chuẩn mực cho giải pháp có thể chấp nhận, và
- nhu cầu đối với hành động phòng ngừa và khắc phục.
11. Hồ sơ kế hoạch hành động phòng ngừa và hành động khắc phục
Tất cả các hành động phòng ngừa, hành động khắc phục, sự không phù hợp, lỗi và sự cố phòng thí nghiệm cần được ghi lại trong hồ sơ kế hoạch hành động phòng ngừa và hành động khắc phục. Hồ sơ cần được duy trì trong sổ nhật ký tổng thể về kế hoạch hành động phòng ngừa và khắc phục.
Các hồ sơ được biên soạn cần được xem xét thường xuyên như một phần của việc xem xét của lãnh đạo.
12. Kế hoạch cải tiến liên tục
Ngay khi việc điều tra hoàn thành, lãnh đạo phòng thí nghiệm cần xem xét thông tin thu được về sự không phù hợp, lỗi và sự cố phòng thí nghiệm. Thông tin này cần được đánh giá về khả năng thích hợp đối với bệnh nhân và sự an toàn của phòng thí nghiệm, đặc biệt với những điều sau:
- có các mối nguy chưa được ghi nhận trước đang tồn tại không,
- có các đánh giá ban đầu sự không phù hợp, lỗi và sự cố phòng thí nghiệm bị làm mất hiệu lực như một kết quả không.
Nếu một trong các điều trên xảy ra, kết quả đánh giá cần được phản hồi lại như một đầu vào trong quá trình đánh giá.
Ngoài ra, một cuộc điều tra chuyên sâu về nguyên nhân gốc rễ của sự không phù hợp, lỗi và sự cố phòng thí nghiệm có nguy cơ cao phải được tiến hành ngay lập tức, nhằm ngăn ngừa sự tái diễn của chúng.
PHỤ LỤC A
(Tham khảo)
Phân tích phương thức và tác động sai lỗi
Phân tích phương thức và tác động sai lỗi (FMEA) là một phương pháp luận để nhận biết các sai sót tiềm ẩn trong một quá trình, nhằm xác định tác động của chúng và nhận biết các hành động để giảm thiểu sai sót. FMEA đặc biệt hữu ích khi quyết định liệu có nên đưa vào phòng thí nghiệm một quá trình mới.
Trong khi khó biết trước từng phương thức sai lỗi, nhóm tham gia nghiên cứu phòng thí nghiệm có thể xây dựng một danh sách các phương thức sai lỗi tiềm ẩn đầy đủ nhất có thể.
Sơ đồ khối của sản phẩm/quá trình chỉ ra các bước chính của quá trình cần được xây dựng. Các bước chính của quá trình phải được liên kết với nhau bằng các đường để chỉ ra các thành phần hoặc các bước liên quan đến nhau như thế nào. Sơ đồ này cho thấy quan hệ lô gic của các thành phần và thiết lập một cấu trúc mà xung quanh cấu trúc này FMEA có thể được xây dựng.
Hình A.1 đưa ra một bản đồ quá trình điển hình trong phòng thí nghiệm y tế, trong đó bao gồm tiền phân tích, phân tích và xử lý sau phân tích mẫu, thuốc thử, thiết bị, dụng cụ,thiết bị hiệu chuẩn,bộ điều chỉnh, trình bày kết quả và tài liệu kết quả.

Hình A.1- Ví dụ về bản đồ quá trình trong phòng thí nghiệm y tế
Phương thức sai lỗi được định nghĩa là cách thức theo đó quá trình có khả năng sai lỗi, cần được nhận biết theo một cách thức có thể xác định tác động cuối cùng sẽ là gì. Tác động của sai lỗi được định nghĩa là kết quả của phương thức sai lỗi thuộc quá trình, như được nhận thấy bởi lỗi của phép thử. Nó được miêu tả theo những gì bệnh nhân có thể trải qua khi nhận ra dạng sai lỗi được nhận biết, ví dụ như sự bất tiện hoặc thiệt hại xảy ra từ kết quả xét nghiệm chẩn đoán hay điều trị chậm trễ hoặc thiếu chính xác.
Một phương thức sai lỗi, trong một thành phần có thể xem là nguyên nhân của phương thức sai lỗi khác trong một hành động khác của quá trình.
Đối với mỗi phương thức sai lỗi được nhận biết, nhóm nghiên cứu cần phải xác định tác động cuối cùng sẽ là gì và hình thành bảng xếp hạng mức độ nghiêm trọng của tác động, nhằm xác định xem sai lỗi nào cần giải quyết trước.
Nhóm nghiên cứu sau đó xác định bộ điều chỉnh và các quy trình giám sát tiềm năng khác để có thể ngăn ngừa nguyên nhân của phương thức sai lỗi xảy ra. Mỗi quy trình cần được đánh giá để xác định nó có thể loại bỏ phương thức sai lỗi tốt thế nào.
Ngay khi một quá trình mới đi vào hoạt động, những phương thức sai lỗi chưa được phát hiện hoặc xác định trước đó có thể xuất hiện. FMEA sau đó cần được cập nhật và các kế hoạch được thực hiện để xác định các sai lỗi đó nhằm loại bỏ chúng khỏi sản phẩm/quá trình.
PHỤ LỤC B
(Tham khảo)
Mô hình đánh giá rủi ro thiệt hại
Hình B.1 trình bày mô hình rủi ro cần được tính toán để đánh giá rủi ro thiệt hại gây ra cho bệnh nhân từ các kết quả sai lầm.
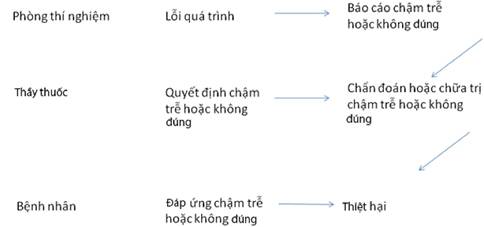
Hình B.1- Ví dụ về mô hình đánh giá rủi ro thiệt hại
Theo mô hình này, hệ quả của việc cung cấp kết quả sai cho bác sỹ sẽ phụ thuộc vào hành động của bác sỹ và ý nghĩa y tế của quá trình phòng thí nghiệm tương ứng. Các bác sỹ dùng kết quả kiểm tra của phòng thí nghiệm cùng với thông tin y tế có sẵn khác để đánh giá bệnh nhân và đi đến quyết định. Trong một vài trường hợp, kết quả của phòng thí nghiệm có thể là cơ sở duy nhất cho quyết định y tế. Xác suất của bệnh nhân bị tổn hại là sự kết hợp của các xác suất mà từng sự kiện được trình bày trong mô hình sẽ xảy ra. Mỗi xác suất riêng biệt được bù một phần bởi xác suất mà tình huống lỗi, rủi ro hay nguy hiểm sẽ được công nhận bởi phòng thí nghiệm hay bác sỹ, từ đó cho phép can thiệp và phòng tránh nguy hại. Chuỗi thực tế của sự kiện sẽ phụ thuộc vào quá trình phòng thí nghiệm cụ thể, và rủi ro từ mỗi sự kết hơp các sự kiện cần được đánh giá độc lập, bằng cách sử dụng sơ đồ này như một hướng dẫn.
PHỤ LỤC C
(Tham khảo)
Xếp hạng mức độ nghiêm trọng
Đáp ứng đối với sai lỗi của phòng thí nghiệm cần phù hợp với cả khả năng tái diễn và mức độ nghiêm trọng tiềm ẩn của kết quả. Nguy cơ lỗi không đáng kể hay hậu quả nhỏ là có thể chấp nhận được, trừ khi chúng có khả năng tái diễn thường xuyên. Mặt khác, nguy cơ lỗi có khả năng đe dọa tính mạng, kể cả khi khả năng xảy ra là hiếm thì cũng không thể chấp nhận được. Trường hơp sự loại bỏ hoặc giảm đủ mối nguy là không thể, thông tin về người nhận kết quả của sự nguy hiểm còn sót lại có thể được cân nhắc như một biện pháp giảm rủi ro. Bảng C.1 trình bày một ví dụ về sự liên kết thường xuyên của rủi ro và kết quả.
Bảng C.1- Ví dụ về xếp hạng mức độ nghiêm trọng liên kết thường xuyên của rủi ro và kết quả
|
Xác suất |
Xếp hạng nghiêm trọng |
||||
|
Không đáng kể |
Nhỏ |
Nghiêm trọng |
Nguy hiểm |
Rất nguy hiểm |
|
|
Thường xuyên |
Không chấp nhận |
Không chấp nhận |
Không chấp nhận |
Không chấp nhận |
Không chấp nhận |
|
Có thể xảy ra |
Chấp nhận |
Không chấp nhận |
Không chấp nhận |
Không chấp nhận |
Không chấp nhận |
|
Thỉnh thoảng |
Chấp nhận |
Chấp nhận |
Chấp nhận |
Không chấp nhận |
Không chấp nhận |
|
Rất ít |
Chấp nhận |
Chấp nhận |
Chấp nhận |
Không chấp nhận |
Không chấp nhận |
|
Không chắc xảy ra |
Chấp nhận |
Chấp nhận |
Chấp nhận |
Chấp nhận |
Chấp nhận |
Thư mục tài liệu tham khảo
[1] ASTION, ML, SHOJANIA, KG, HAMILL, TR, KIM, S and NG, VL, “Classifying laboratory incident reports to identify problems that jeopardize patient safety”, American Journal of Clinical Pathology (2003), 120 (1), pp. 18-26 (Phân loại báo cáo sự cố phòng thí nghiệm để xác định các vấn đề gây nguy hiểm cho sự an toàn của bệnh nhân)
[2] KOHN, LT, CORRIGAN, JM and DONALDSON, MS, eds. To Err is Human: Building a Safer Health System, Institute of Medicine, Washington DC: National Academy Press (2000), (3) (Xây dựng hệ thống y tế an toàn)
[3] SPATH, PL, “Using failure mode and effects analysis to improve patient safety”, Association of Operating Room Nurses (AORN) Journal(2003), 78 (2), pp. 16-37 (Sử dụng phân tích phương thức và tác động sai lỗi để cải thiện an toàn cho bệnh nhân)
[4] BONINI, PA, PLEBANI, M, CERIOTTI, F and RUBBOLI, T, “Errors in laboratory medicine”, Clin Chem. (2002), 48, pp. 691-698 (Lỗi trong phòng thí nghiệm y tế)
[5] CLSI/NCCLS, A Quality Management System Model for Health Care; Approved Guideline (2nd Edition), NCCLS document HS1-A3, Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA (2004) (Mô hình hệ thống quản lý chất lượng cho việc chăm sóc sức khỏe)
[6] CLSI/NCCLS, Continuous Quality Improvement: Integrating Five Key Quality System Essentials; Approved Guideline (2nd Edition), NCCLS document GP22-A2, Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA (2004) (Cải tiến liên tục chất lượng: Tích hợp năm yếu tố của hệ thống chất lượng)
[7] CLSI/NCCLS, A Quality Management System Model for Health Care; Approved Guideline (2nd Edition), NCCLS document HS1-A3, Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA (2004) (Mô hình hệ thống quản lý chất lượng cho việc chăm sóc sức khỏe)
[8] CLSI/NCCLS, Quality Management for Unit-Use Testing; Approved Guideline, NCCLS document EP18-A, Clinical and Laboratory Standards Institute, 940 West Valley Road, Suite 1400, Wayne, Pennsylvania 19087-1898 USA (2002) (Quản lý chất lượng cho các đơn vị sử dụng thử nghiệm)
[9] REASON, JT, Human Error, New York, NY: Cambridge University Press (1990) (Lỗi của con người)
[10] DAVIDSONFRAME, J, Managing Risk in Organizations: A Guide for Managers, San Francisco CA: Jossey-Bass (2003) (Quản lý rủi ro trong các tổ chức: Hướng dẫn cho người quản lý)
[11] ROBITAILLE, D, The Preventive Action Handbook, Chico, CA: Paton Press (2003) (Sổ tay hành động phòng ngừa)
[12] Guidelines for Hazard Evaluation Procedures — With Worked Examples (2nd Edition), Center for Chemical Process Safety/AIChE (1992), ISBN 0-8169-0491-X (Hướng dẫn thủ tục đánh giá mối nguy – Ví dụ làm việc)
[13] MIKE, W. Schmidt, The Use and Misuse of FMEA in Risk Analysis, (MDDI March 2004) (Sử dụng và sử dụng sai FMEA trong phân tích rủi ro)
[14] STAMATIS, D.H., Failure Mode and Effect Analysis: FMEA From Theory to Execution (2nd Edition, 2003), ISBN 0873895983 (Phân tích phương thức và tác động sai lỗi: FMEA từ lý thuyết đến thực hành)
[15] MIL-STD-1629A, Military Standard Procedures for Performing a Failure Mode, Effects and Criticality Analysis, United States Department of Defense, Washington, DC 20301 (1980) (Thủ tục tiêu chuẩn quân sự đối với việc thực hiện phân tích phương thức, tác động sai lỗi)
[16] IEC 60812, Analysis techniques for system reliability — Procedure for failure mode and effects analysis (FMEA) (Kỹ thuật phân tích tính tin cậy của hệ thống – Quy trình phân tích phương thức và tác động của sai lỗi)
[17] IEC 61025, Fault tree analysis (FTA) (Phân tích cây lỗi)
[18] IEC 61882, Hazard and operability studies (HAZOP studies) - Application guide (Nghiên cứu mối nguy và khả năng hoạt động– Hướng dẫn áp dụng)
MỤC LỤC
Lời nói đầu
Lời giới thiệu
1. Phạm vi áp dụng
2. Tài liệu viện dẫn
3. Thuật ngữ và định nghĩa
4. Trách nhiệm của lãnh đạo đối với hành động khắc phục, phòng ngừa và cải tiến liên tục
5. Nhận biết sự không phù hợp, lỗi và sự cố thực tế và tiềm ẩn của phòng thí nghiệm
6. Phân loại sự không phù hợp, lỗi và sự cố của phòng thí nghiệm
7. Hành động phòng ngừa và hành động khắc phục
8. Đánh giá rủi ro nảy sinh từ sự không phù hợp thực tế và tiềm ẩn của phòng thí nghiệm
9. Xem xét sự không phù hợp, lỗi và sự cố phòng thí nghiệm thu thập được
10. Kế hoạch hành động phòng ngừa và hành động khắc phục
11. Hồ sơ kế hoạch hành động phòng ngừa và hành động khắc phục
12. Kế hoạch cải tiến liên tục
Phụ lục A (tham khảo) Phân tích phương thức và tác động sai lỗi
Phụ lục B (tham khảo) Mô hình đánh giá rủi ro thiệt hại
Phụ lục C (tham khảo) Xếp hạng mức độ nghiêm trọng
Thư mục tài liệu tham khảo


