Tiêu chuẩn Việt Nam TCVN 6470:1998 (Phần IV JECFA - FAO FOOD and nutrition paper - 5 Rev. 2) về phụ gia thực phẩm - Phương pháp xác định cho phẩm màu thực phẩm do Bộ Khoa học Công nghệ và Môi trường ban hành đã được thay thế bởi Tiêu chuẩn quốc gia TCVN 6470:2010 về Phụ gia thực phẩm - Phương pháp thử đối với các chất tạo màu .
Nội dung toàn văn Tiêu chuẩn Việt Nam TCVN 6470:1998 (Phần IV JECFA - FAO FOOD and nutrition paper - 5 Rev. 2) về phụ gia thực phẩm - Phương pháp xác định cho phẩm màu thực phẩm do Bộ Khoa học Công nghệ và Môi trường ban hành
TIÊU CHUẨN VIỆT NAM
TCVN 6470 : 1998
PHỤ
GIA THỰC PHẨM - PHƯƠNG PHÁP XÁC ĐỊNH CHO PHẨM MÀU THỰC PHẨM
Food additives - Methods for food colours
Lời nói đầu
TCVN 6470 : 1998 hoàn toàn phù hợp với phần IV của sách Hướng dẫn yêu cầu kỹ thuật cho những chú ý chung, thử nhận biết, dung dịch thử và các tài liệu tham khảo khác của JECFA (Guide to specifications, general notices, general analytical techniques, identification tests, test solutions and other reference materials - JECFA - FAO FOOD and nutrition paper - 5 rev 2).
TCVN 6470 : 1998 do ban kỹ thuật tiêu chuẩn TCVN/TC/F4 Phụ gia thực phẩm biên soạn, Tổng cục Tiêu chuẩn - Đo lường - Chất lượng đề nghị, Bộ Khoa học, Công nghệ và Môi trường ban hành.
PHỤ GIA THỰC PHẨM - PHƯƠNG PHÁP XÁC ĐỊNH CHO PHẨM MÀU THỰC PHẨM
Food additives - Methods for food colours
Tiêu chuẩn này qui định phương pháp xác định cho phẩm màu thực phẩm.
1 Xác định clorua theo natri clorua
1.1 Thiết bị
Thiết bị chuẩn độ điện thế, với điện cực chỉ định bạc, điện cực so sánh calomel, và cầu nối la dung dịch kali sunfat bão hoà.
1.2 Cách tiến hành
Cân 0,5 - 1,0 g mẫu phẩm màu, hoà tan trong 100 ml nước, và axit hóa bằng 5 ml dung dịch axit nitric 1,5 N. Đặt điện cực bạc vào dung dịch màu và nối điện cực calomel với dung dịch bằng cầu nối kali sunfat bão hoà. Có thể loại bỏ cầu nối kali sunfat bão hoà bằng cách dùng một điện cực thuỷ tinh làm điện cực so sánh. Điều này là đơn giản thiết bị một cách đáng kể và điện cực thuỷ tinh này đủ ổn định để được sử dụng như một điện cực so sánh cho kiểu chuẩn độ này.
Xác định hàm lượng clorua của dung dịch bằng chuẩn độ với dung dịch bạc nitrat 0,1 N, và tính toán kết quả theo natri clorua, 1 ml dung dịch bạc nitrat 0,1 N = 0,00585 g natri clorua.
Biểu thị kết quả bằng phần trăm của lượng mẫu đã lấy.
Chú thích - Nhiệt độ qui định ở tiêu chuẩn này là độ C (0C).
2 Các chất không tan trong clorofooc
2.1 Thiết bị
- tủ sấy, dải nhiệt độ: 00 - 2000;
- bếp điện, đun sôi được tetraclorua cacbon (CCl4) (điểm sôi 76,80);
- chén nung, có lớp lọc bằng bông thuỷ tinh;
- bình hút chân không;
- máy hút chân không;
- bình hút ẩm.
2.2 Thuốc thử
- tetraclorua cacbon, loại thuốc thử, hay;
- clorofooc, loại thuốc thử;
(Các loại thuốc thử này được gọi là "dung môi" trong phần "cách tiến hành").
2.3 Cách tiến hành
Tiến hành thử theo cách sau: Trộn lượng mẫu cân theo yêu cầu (W1) với 100 ml dung môi trong cốc có mỏ dung tích 250 ml, khuấy đều, đun đến sôi trên bếp điện trong tủ hút (fume hood). Lọc dung dịch nóng trên vào 1 chén nung đã cân trước (W2). Chuyển cặn trong cốc có mỏ vào chén nung bằng dung môi. Rửa cặn trong chén nung với từng phần 10 ml dung môi cho đến khi nước rửa không màu. Đặt chén nung này vào tủ sấy và sấy ở 1000 - 1500 trong 3 giờ. Để nguội chén nung trong bình hút ẩm. Cân chén nung đã nguội (W3).
2.4 Tính toán
Tính phần trăm chất không hoà tan trong tetraclorua cacbon hay clorofooc (PIM) trong mẫu, theo công thức sau:
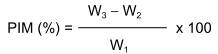
Tính kết quả là phần trăm chất không tan trong tetraclorua cacbon (CCl4) hay clorofooc có trong mẫu phân tích.
3 Các chất màu thực phẩm
3.1 Nhận biết
Nhiều chất màu thực phẩm được sử dụng trong công nghiệp chế biến thực phẩm ở dạng hỗn hợp các chất màu thực phẩm như mô tả trong các chuyên luận, đôi khi trong hỗn hợp có thêm chất làm loãng.
Tiến hành phép thử đơn giản để xem liệu mẫu bột là một chất màu đơn, hay là một khối trộn vật lý của một số phẩm màu. Rải một lượng bột màu rất nhỏ vào 2 cốc, một cốc chứa nước, một cốc khác chứa axit sunfuric đặc. Dưới điều kiện này những hạt nhỏ li ti của các chất màu riêng rẽ có thể nhìn thấy dễ dàng khi chúng hoà tan và, phép thử này có độ nhậy bất ngờ.
Việc xác định sự có mặt các chất màu thực phẩm riêng biệt thường khó khăn. Một số lượng lớn các muối natri sunfonic và kết quả là chúng không có điểm chảy và điểm sôi chính xác. Thêm vào đó các chất màu tổng hợp thường có chứa các chất màu phụ trong khi các chất màu được chiết xuất từ nguồn thiên nhiên nhìn chung có chứa một số màu cơ bản khác nhau. Do đó việc nhận biết thành công nhất bằng cách so sánh các tính chất quan sát được với các đặc tính của mẫu thương phẩm thật.
Các kĩ thuật chính được sử dụng là sắc kí và quang phổ, hoặc kết hợp 2 phương pháp. Ví dụ khi có mặt các chất màu phụ có thể ảnh hưởng tới phổ quan sát và việc xác định sự có mặt các thành phần chính không thể thực hiện được. Vì lý do đó, nên tách các chất màu bằng sắc kí cột, sắc kí giấy hay sắc kí lớp mỏng trước khi định dùng các phương pháp nhận biết khác.
Sắc kí giấy và sắc kí lớp mỏng thường rất hữu ích trong việc nhận biết các chất màu, mà không cần đến các thiết bị đắt tiền. Nhưng luôn phải nhớ rằng giá trị R4 của một chất chỉ khẳng định bằng lý thuyết. Trong thực tế hầu hết được dựa trên quá trình kiểm tra. Có nhiều yếu tố ảnh hưởng, làm cho giá trị Rf trở nên rất không ổn định. Các yếu tố đó bao gồm: thành phần và thời hạn (tuổi) của dung môi, nồng độ của dung môi bốc hơi trong không khí, chất lượng giấy, máy móc dụng cụ, loại và chất lượng của các chất phụ, nồng độ, giá trị pH của dung dịch và nhiệt độ. Vì lý do đó, sắc kí so sánh luôn luôn cần sử dụng. Bằng cách cho chạy đồng thời một vài chất với nồng độ như nhau, một số yếu tố ảnh hưởng này sẽ bị loại bỏ.
Sự trùng hợp về khoảng di chuyển với một hệ dung môi đơn phải được coi như là một chỉ tiêu nhận biết và các phép thử khác sẽ phải được tiến hành để khẳng định chắc chắn. Bảng sau đây cho những thí dụ về giá trị Rf của dung dịch 1 % các chất màu trong nước, triển khai trên sắc kí lớp mỏng silicagel G tại 10 hệ dung môi khác nhau. Thành phần của các hệ thống dung môi này, (tất cả phải được chuẩn bị mới) - là:
Dung môi No:
1. lso - Propanol: amonac (tỷ trọng 0,880): nước (7 : 2 : 1)
2. lso - Butanol: etanol: nước: amoniac (tỷ trọng 0,880) (10 : 20 : 10 : 1)
3. Dung dịch kali nitrat bão hoà trong nước
4. Phenol: nước (4 : 1, W/v)
5. Axit clohydric (tỷ trọng 1,180): nước (23: 77)
6. Trinatrixitrat: amoniac (tỷ trọng 0,880): nước (2 g : 15 ml : 85 ml)
7. Axeton: etylmetyl ketone: amoniac (tỷ trọng 0,880): nước (60 : 140 : 1 : 60)
8. n - Butanol: etanol: pyridin: nước (2 : 1 : 1 : 2)
9. lso - propanol: amoniac (tỷ trọng 0,880) (4 : 1)
10. n - Butanol: axetic axit (băng): nước (10 : 5 : 6)
Việc đánh giá các vết màu cần làm trong khi sắc đồ hãy còn ẩm dung môi, và cả sau khi sấy khô. Các vết màu sẽ được xem xét dưới tia sáng trắng tới và xuyên qua, tốt hơn như trong ánh sáng tử ngoại. Dưới ánh sáng tử ngoại (UV) nhiều chất màu có đặc tính thay đổi màu. Hơn nữa điều này vì thế thường có thể có đối với các vết tạp chất huỳnh quang không màu. Nếu có thể, sử dụng hai bức xạ UV với các chiều dài sóng hiệu xuất khác nhau; một đèn phát xạ xung quanh 250 nm.
Nên tiến hành các phép thử đối với axit, kiềm và thuốc thử thích hợp khác, để đảm bảo kết quả đúng. Tất cả các phép thử có thể được tiến hành với pipet mao quản trên mỗi vết màu.
Các yêu cầu sau đây phải đạt được khi xác định các chất màu:
- các khoảng di chuyển ngang bằng trong một vài dung môi;
- các vết ngang bằng dưới ánh sáng thường và ánh sáng tử ngoại;
- sự thay đổi màu tương đương với các thuốc thử.
Việc kiểm tra bằng phương pháp quang phổ là phương pháp tốt nhất để xác định các chất màu. Các vùng tử ngoại, hồng ngoại và ánh sáng nhìn thấy được đều được áp dụng.
Vùng nhìn thấy được của quang phổ nhìn thấy được thông thường là phép kiểm tra bước đầu tiên trong việc thử nhận biết một chất màu chưa biết. Có nhiều chất màu cho độ hấp thụ đặc trưng trong vùng nhìn thấy được, trong khi các chất màu khác thì không. Phổ trong vùng tử ngoại cũng có thể được sử dụng, và nên thu nhận kết quả này, nếu có thể được.
Phổ hấp thụ hồng ngoại thường là cách tốt nhất để xác định các hợp chất khác nhau, nhưng có một số khó khăn trong quá trình sử dụng phương pháp này.
Trong việc áp dụng phép đo quang phổ nhìn thấy được và tử ngoại, phổ cần phải luôn thu nhận được trong một vài dung môi, hay nếu trong dung môi riêng lẻ, thì phải dưới những điều kiện khác nhau.
Phổ của dung dịch nước cần thu được trong những điều kiện trung tính (đệm bằng amoni axetat), axit (axit clohidric 0,1 N), và kiềm (natri hydroxit 0,1 N).
Những giá trị Rf của một sô phẩm màu được phép và không được phép tan trong nước
|
Tên phẩm màu |
c 1 N° |
EEC Seral M No |
Dung môi số (xem mục 3-1.) |
|||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|||
|
Màu đỏ Ponceau 4 R hay cochineal Red A |
16255 |
E 124 |
0,66 (0,85) |
0,75 |
0,88 |
0,03 |
0,95 |
1,00 |
0,60 |
0,90 |
0,11 |
0,52 (0,00-057 |
|
Carmoisine hay Azorubine |
14720 |
E 122 |
0,65 (0,77) |
0,81 |
0,00-0,42 |
0,16 |
0,00 (0,00-0,32) |
1,00 |
0,65 |
0,88 |
0,34 (0,46) |
0,63 (0,11-0.70) |
|
Amaranth |
16185 |
E 123 |
0,62 (0,48-0,76) |
0,75 (0,83) |
1,00 (0,00-1,00) |
0,04 (0,16) |
1,00 |
1,00 |
0,40 (0,64-0,66) |
0,90 |
0,10 (0,41) |
0,39-0,67 |
|
Erythrosine Rs |
45430 |
E 127 |
0,85 (0,68-0,79) |
0,91 (0,86-0,74-0,81) |
0,00-0,10 |
0,00-0,90 (0,41) |
0,00 |
0,00-0,95 |
0,64-0,66 (0,58) |
0,89 |
0,66 (0,57-0,43) |
1,00 |
|
Red 2 G Màu vànq cam |
18050 |
|
0,68 |
0,80 |
0,37 |
0,12 |
0,00-0,71 |
1,00 |
0,64 |
0,90 |
0,36 |
0,68 |
|
Orange G |
16230 |
|
0,71 (0,67-0,88) |
0,80 (0,75) |
0,64 (1,00-0,35) |
0,23-0,15 0,04 |
0,73 |
1,00 |
0,64 (0,62-0,50- 0,67) |
0,91 |
0,36 (0,32-0,17) |
0,69 (0,46-0,82) |
|
Orange R.N |
15970 |
|
0,83 (0,62) |
0,88 (0,78) |
0,00 (0,00-0,42) |
0,42 (0,13) |
0,13 (0,38) |
0,76 (1,00) |
0,68 (0,65) |
0,92 |
0,64 (0,29) |
0,82-0,71 |
|
Sunset yellovv FCF hay Orange yellovv s Màu vànq |
15985 |
E 110 |
0,75 (0,68) |
0,82 (0,74) |
1,00 (0,00-1,00) |
0,17-0,03 |
1,00 |
1,00 |
0,65 (0,48) |
0,90 |
0,34 (0,10-0,22) |
0,67 (0,46) |
|
Tatrazine |
19140 |
E 102 |
0,66 |
0,77 |
0,46-1,00 |
0,08 |
1,00 |
1,00 |
0,52 |
0,93 |
0,14 |
0,50 |
|
Yellovv 2 G |
18965 |
|
0,63 |
0,80 |
0,77 |
0,21 |
0,74 |
1,00 |
0,62 |
0,92 |
0,21 |
0,75 |
|
Quinoline Yellovv |
47005 |
E 104 |
0,83-0,88 |
0,88 (0,82) |
0,00-1,00 |
0,65 (0,21) |
0,26-1,00 0,00-0,38 |
0,95 (0,35) |
0,54 (0,68) |
0,88 |
0,00-0,31 0,64 |
0,11-0,75 (0,83) |
|
Fast yellovv AB Màu xanh |
13015 |
E 105 |
0,77 |
0,81 |
1,00 |
0,14 |
0,97 |
1,00 |
0,56 |
0,93 |
0,36 |
0,66 |
|
Green s hay axit Brillant green BS hay Lissamine green |
44090 |
E 142 |
0,44 (0,52-0,68 0,74) |
0,61 (0,67-0,75- 0,81-0,84) |
0,49 (0,24) |
0,52 (0,05-0,36 1,00) |
0,29 (0,43) |
1,00 40,94 |
0,46 (0,56-0,71) |
0,75 (0,89-0,92) |
0,07 |
0,55 |
|
Indigo Carmine hay Indigotin Indanthrene Blue hay |
73015 |
E 132 |
0,56 (0,70) |
0,50-0,76 (0,78) |
0,00 (0,05-0,90 1,00) |
0,09-0,18 (0,52) |
0,92 |
|
0,66 (0,71-0,73) |
0,89-0,84 |
0,37 (0,00-0,34) |
0,00-0,63 |
|
Solanthrene Blue RS hay Anthragen Blue |
69800 |
E 130 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
0,00 |
Beillant blue FCF |
42090 |
|
0,64 |
0,78 |
0,05 |
0,45 |
0,10 |
0,00-1,00 |
0,61 |
0,88 |
0,30 |
0,53 |
|
|
|
|
(0,73) |
|
|
(0,68) |
|
|
(0,68) |
|
(0,49-0,00-0,23) |
(0,64) |
|
Patent Blue V |
42051 |
E131 |
0,34-0,60 |
0,68 |
0,05 |
0,55 |
0,15 |
0,95 |
0,69 |
0,84 |
0,00-0,10 |
0,59 |
|
|
|
|
|
|
|
|
|
|
(0,72) |
(0,92) |
|
|
|
Violet 6B |
42640 |
|
0,73 |
0,80 |
0,00 |
0,62 |
0,00-0,37 |
0,00-1,00 |
0,67 |
0,89 |
0,37-0,45 |
0,64 |
|
|
|
|
(0,67-0,91) |
(0,72) |
(0,00-0,46) |
(0,51-1,00) |
|
|
|
(0,62) |
0,70-0,76 |
(0,71 |
|
Methyl Violet |
42535 |
|
0,91 |
0,56-0,81 |
0,00 |
0,79-1,00 |
0,00-0,80 |
0,00 |
0,00-0,68 |
0,11 |
0,94-0,87 |
0,75-0,79) |
|
|
|
|
(0,80) |
0,90 |
(0,00-0,34) |
|
|
(0,00-0,53) |
(0,11-0,28 |
(0,90) |
(0,000,83) |
(0,00-0,70) |
|
Màu nâu, đen |
|
|
|
|
|
|
|
|
0,53-1,00) |
|
|
|
|
Brovvn FK |
|
|
0,78-0,71 |
0,79-0,86 |
1,00 |
0,69-0,27 |
0,00-0,77 |
1,00 |
0,59-0,64 |
0,93 |
0,34 |
0,00-0,73 |
|
|
|
|
0,66 |
|
(0,00-1,00) |
0,15-0,00 |
|
|
0,53 (0,37) |
|
(0,26-0,53) |
|
|
Chocolate Brovvn FB |
|
|
0,00-0,69 |
0,00-0,75 |
0,00-0,82 |
0,00 |
0,00-1,00 |
0,00-1,00 |
0,36-0,51 |
0,87 |
0,00-0,38 |
0,00-0,75 |
|
|
|
|
|
|
|
(0,00-0,23) |
|
|
0,62 |
|
|
|
|
Chocolate Brovvn HT |
20285 |
|
0,00-0,63 |
0,74 |
0,00-1,00 |
0,00 |
0,00-1,00 |
0,00-1,00 |
0,34-0,43 |
0,88 |
0,00-0,32 |
0,00-0,73 |
|
|
|
|
|
|
|
(0,00-0,16) |
|
|
0,62 |
|
|
|
|
Black PN hay |
28440 |
E151 |
0,66 |
0,75 |
1,00 |
0,00 |
1,00 |
1,00 |
0,38 |
0,85 |
0,05 |
0,00-0,43 |
|
Brillant - Black BN |
|
|
(0,47) |
|
(0,00-1,00) |
|
|
|
(0,61) |
|
|
|
|
Black 7984 |
27755 |
E152 |
0,62 |
0,75 |
1,00 |
0,00 |
1,00 |
1,00 |
0,38 |
0,85 |
0,09 |
0,00-0,45 |
|
|
|
|
|
|
|
|
|
|
(0,61) |
|
|
|
Cl. No: số chỉ sô' phẩm màu" con số trong ngoặc chỉ các vết phụ của cường độ thấp hơn.
O.x - O . y: kẻ vạch giữa các vết.
Theo Pearson, D (1973), J.ASSOC Public. Anal, 11,137-138 tái bản theo chất sinh ra từ ung thư do môi trường. Phương pháp phân tích tuyển chọn, tập 4 - Một chất Amin thơm và thuốc nhuộm.
A30 trong tài liệu môi trường chung và môi trường công nghiệp (ấn phẩm IARC số 40) chi nhánh quốc tế về nghiên cứu ung thư, Lyon, 1981.
Phổ hấp thụ được dựng thành những đồ thị cho thấy độ hấp thụ ở mọi bước sóng. Việc kiểm tra các đường cong kết quả bao gồm nhiều vùng của bước sóng có độ hấp thụ cực đại. Toàn bộ đường cong cần được xem xét cận thẩn để xác định dạng riêng biệt của đường cong từ "các gờ" hay các điểm uốn có thể là các nét đặc trưng và hữu ích nhất của phổ hấp thụ. Những nét đặc trưng này thường có thể phân biệt được giữa hai hoặc nhiều các chất màu có độ hấp thụ tối đa ở cùng bước sóng. Nhiều chất màu có thể xác định bằng việc quan sát phạm vi mà độ hấp thụ cực đại và những nét đặc trưng khác của đường cong hấp thụ bị thay đổi trong các điều kiện pH khác nhau hoặc bởi có thay đổi khác nhau trong dung môi.
Phổ hồng ngoại có thể thu nhận được bằng một vài cách; những cách thông dụng là:
- phổ của dung dịch chất thử trong các dung môi thích hợp;
- phổ của chất huyền phù trong 1 chất lỏng thích hợp.
- kĩ thuật tạo viên kali bromua (một lượng nhỏ chất màu, thường từ 1 - 3 mg được trộn đều, kĩ với kali bromua khô, tinh khiết, hỗn hợp này được chuyển thành những viên như hạt lựu thích hợp và được ép thành dạng cứng bằng cách dùng áp suất nén từ 700 đến 1400 kg/cm2).
Dựng đồ thị phổ thu được theo cách thông thường. Những nét đặc trưng nổi bật của các đường cong kết quả là các bước sóng tại đó các đỉnh (Pic) hấp thụ và dạng của các đường cong gần kề những Pic đó.
Việc thảo luận chi tiết để giải thích phổ hồng ngoại ngoài phạm vi của tiêu chuẩn này. Tuy nhiên, cần phải thận trọng để đảm bảo chắc chắn rằng các Pic hấp thụ do các chất tạp nhiễm vô cơ thì không được coi là do chất màu.
Cấu trúc tinh thể hoặc trạng thái vật lí khác của mẫu thử có thể ảnh hưởng đến phổ thu được từ huyền phù hay các viên kali bromua. Cần đảm bảo chắc chắn rằng các chất chưa biết sẽ được xử lý chính xác theo cùng cách thức như chất chuẩn hay mẫu đã biết. Các chất màu tan trong nước, thường có thể được xử lý bằng cách hoà tan chúng trong nước, thêm một ít axit axetic, cho bốc hơi (cô) đến gần khô, và sau đó sấy khô ở khoảng 1000 để loại hết nước còn dư. Tất cả các chất màu được kiểm tra phải không còn nước hoặc dung môi khác trước khi thu nhận phổ hồng ngoại. Nước dung môi hữu cơ hấp thụ bức xạ hồng ngoại.
Một thí dụ về việc sử dụng hồng ngoại, có hai chất màu, chất có liệt kê trong tiêu chuẩn này, có thể được để cập đến Phẩm màu Sunset yellow và phẩm màu Orange GGN có phổ hấp thụ trong vùng nhìn thấy được và vùng tử ngoại gần giống nhau đến nỗi mà chúng không thể phân biệt được qua việc kiểm tra ở những vùng này. Tuy nhiên, phổ hồng ngoại của chúng, hoàn toàn khác trong vùng phổ mà nhóm axit sunfonic hấp thu rất mạnh.
Trong một vài trường hợp, quy trình sắc kí, qui trình quang phổ, và bất kì một sự kết hợp của hai quy trình trên có thể không cho kết quả đúng. Trong những trường hợp như vậy, vấn đề thường được giải quyết bằng cách giảm bớt chất màu bằng cách khác giảm các hợp chất này và xác định các sản phẩm tạo ra. Kỹ thuật này đặc biệt được áp dụng cho các chất màu azot. Các hợp chất amin tạo ra từ sự phân giảm có thể xác định ngay bằng kĩ thuật sắc kí và đo quang phổ.
Nhiều kĩ thuật khác đã từng được áp dụng để nhận biết các chất màu. Việc mô tả chúng ở đây sẽ không được đề cập, nhưng một ví dụ sẽ được trích dẫn. Nhiều sắc tố có cấu trúc tinh thể nhất định và có thể được nhận biết bằng hình nhiễu xạ X quang, hay bằng quang học tinh thể. Một vài chất màu có thể được chuyển thành dạng dẫn xuất tinh thể và được nhận biết theo cách tương tự.
3.2 Xác định hàm lượng tổng số (tổng hàm lượng)
Có hai phương pháp thường dùng để xác định tổng hàm lượng chất màu: Quang phổ so sánh.
Và Sự phân giảm clorua titan.
Khi sử dụng phương pháp đo quang phổ cần nhớ rằng nhiều nhà sản xuất quang phổ kế không đảm bảo độ chính xác lớn hơn ± 1 %.
Tất cả các chất màu có mặt trong mẫu, mà chúng có Pic hấp thụ nằm trong vùng của chất màu chính, nó sẽ góp phần vào chỉ số hấp thụ để tính toán kết quả. Các chất màu phụ của nhiều màu sắc khác nhau sẽ không được tính đến bằng kĩ thuật này. Trong điều kiện lí tưởng, quang phổ kế được sử dụng để tiến hành phép so sánh giữa mẫu thử và chất chuẩn có hàm lượng chất màu đã biết. Việc sử dụng chỉ số hấp thụ nói chung được chấp nhận thay cho bản thân chất chuẩn, trong chừng mực nào đó được coi như ít thích hợp, nhưng trong trường hợp một số chất màu, được ghi nhận là không thay thế được.
Trong phương pháp titan clorua, giả định rằng các đồng phân và các chất phụ có cùng giá trị đương lượng titan clorua như chất màu chính.
3.3 Xác định hàm lượng tổng số bằng phương pháp quang phổ
Có hai qui trình thực nghiệm được mô tả, chúng khác nhau ở chi tiết và cả hai đều sử dụng số phổ hấp thụ được trích dẫn trong đặc tính chất màu để tính toán kết quả.
Qui trình thứ nhất là điển hình cho dạng sử dụng đối với các chất màu tan trong nước; Qui trình thứ hai thích hợp cho các chất màu tan trong dung môi, đặc biệt với các carotenoit tổng hợp. (Dung dịch được chuẩn bị cho trường hợp thứ hai được dùng trong các phép thử nhận biết carotenoit).
3.3.1 Nguyên tắc
Độ hấp thụ của dung dịch chất màu được xác định tại bước sóng hấp thụ tối đa và được so sánh với độ hấp thụ của chất chuẩn.
3.3.2 Dụng cụ
- máy quang phổ có độ chính xác (± 1 % hay chính xác hơn) và đo độ hấp thụ ở vùng bước sóng 350 nm - 700 nm với độ rộng khe sáng là 10 nm hay nhỏ hơn.
- cuvet có chiều dài 1 cm đường quang.
3.3.3 Thuốc thử
Nước cất mới hoặc dung môi được chỉ định theo đặc tính của chất màu.
3.3.4 Qui trình 1
3.3.4.1 Cân chính xác 0,25 g (± 0,02 g) mẫu, chuyển vào một bình định mức 1000 ml. Thêm nước cất mới vào hoặc dung môi chỉ định và lắc cho hoà tan hết. Làm đầy đến vạch và trộn đều. Pha loãng thành một dung dịch có nồng độ thích hợp tuỳ theo chi tiết đã cho về đặc tính chất màu đó. Xác định độ hấp thụ (A) tại bước sóng hấp thụ tối đa đung cuvet dầy 1 cm.
3.3.4.2 Tính toán:
Tính toán tổng hàm lượng các chất màu của mẫu thử, theo công thức sau:
![]()
trong đó
A là độ hấp thụ của mẫu
A ![]() 1cm là độ hấp thụ đặc trưng của chất chuẩn từ
đặc tính của màu thể tích pha loãng
1cm là độ hấp thụ đặc trưng của chất chuẩn từ
đặc tính của màu thể tích pha loãng
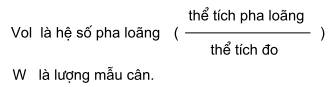
3.3.5 Qui trình 2
3.3.5.1 Cân chính xác khoảng 0,08 g (W) mẫu trong bình định mức 100 ml (V1) và hoà tan bằng cách lắc kĩ với 20 ml dung môi clorofooc tinh khiết, không có axit. Phải làm cho dung dịch trong suốt.
Thêm xyclohexan tính khiết đến vạch. Hút 5,0 ml dung dịch trên (v1) cho vào 1 bình định mức 100 ml (v2) và thêm xyclohexan đến vạch. Tương tự như vậy, pha loãng 5,0 ml dung dịch trên (v2) thành 100 ml (v3) và đo độ hấp thụ tại độ hấp thụ tối đa (A), dùng xyclohexan làm mẫu trắng, sử dụng cuvét 1 cm.
3.3.5.2 Tính hàm lượng tổng số chất màu theo công thức sau:
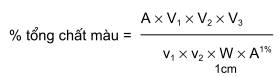
trong đó
A là độ hấp thụ của dung dịch mẫu ở bước sóng có độ hấp thụ tối đa;
V1 là thể tích bình định mức đầu tiên (= 100 ml);
V2 là thể tích bình định mức thứ hai (= 100 ml);
V3 là thể tích bình định mức cuối (= 100 ml);
v1 là thể tích hút pipet lần đầu tiên (5 ml);
v2 là thể tích hút pipet lần thứ hai (5 ml);
W là lượng cân mẫu, tính bằng gam;
A%1cm là độ hấp thụ đặc trưng của chất chuẩn.
Phải hoàn thành việc xác định nhanh chóng, để tránh sự phân huỷ do không khí đến mức có thể, và tiến hành tất cả công đoạn ở nơi không có ánh sáng.
3.3.6 Qui trình xác định phẩm màu đỏ tía (lakes)
Qui trình 1 trên, có thể được áp dụng theo cách sau để xác định tổng hàm lượng chất màu của phẩm màu đỏ tía (lakes).
Chuẩn bị dung dịch đệm photphat pH theo cách sau:
Hoà tan 13,61 g kali hidydrophotphat trong nước và pha loãng đến 1000 ml. Thêm khoảng 90 ml dung dịch NAOH N. Xác định pH bằng pH met và thực hiện việc điều chỉnh dần dần pH đến khi đạt pH = 7,0. Bằng cách sử dụng dung dịch natri hydroxyt khoảng 0,1 N hoặc axit photphoric loãng.
Cân chính xác một lượng phẩm màu đỏ tía, sẽ cho độ hấp thụ xấp xỉ với độ hấp thụ của dung dịch mầu gốc khi tiến hành thử theo qui trình 1 ở trên. Chuyển vào 1 cốc có mỏ chứa 10 ml axit clohydric pha loãng đến khoảng 50 ml với nước. Đun nóng và khuấy để hoà tan phẩm màu đỏ tía (lakes), để nguội đến nhiệt độ phòng và pha loãng đến đúng 1000 ml với dung dịch đệm photphat pH 7. Rồi tiến hành như qui trình 1 ở trên, và trong bảng tra chất màu, dùng dung dịch đệm photphat có pH 7 như một dung môi.
3.3.7 Xác định tổng hàm lượng chất màu bằng phương pháp chuẩn độ với titan clorua
3.3.7.1 Thiết bị, dụng cụ
- máy sinh cacbon dioxit, ví dụ: bộ bình Kipp;
- dụng cụ chuẩn độ, như trong hình 1, bao gồm một bình hút và một buret được nối với một đôi vòi chéo nhau và nhánh bên cạnh. Bình hút được nút chặt miệng bình bằng một nút cao su có hai lỗ, một lỗ thông với ống thuỷ tinh được nối với bình sinh cacbon dioxit, lỗ thứ hai thông với 1 ống thuỷ tinh và được nối với đầu buret. Nhánh bên cạnh của buret được nối với đáy của bình hút;
- bình nón 500 ml có ống dẫn CO2, (khi tiến hành chuẩn độ với titan clorua, phải đảm bảo miệng ống buret luôn luôn ở bên trong cổ của bình nón).
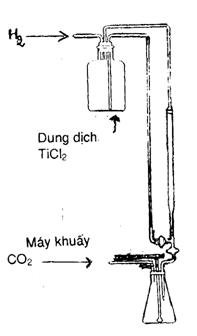
Hình 1 - Dụng cụ để chứa dung dịch titan clorua
3.3.7.2 Cách tiến hành
Chuẩn bị dung dịch titan clorua 0,1 N. Đong vào một bình cầu, lớn, một thể tích dung dịch titan clorua 15 % có chứa 15,5 g - 16,5 g titan clorua trong mỗi lít dung dịch yêu cầu. Thêm 100 ml axit clohydric (tỉ trọng 1,16 - 1,18) cho mỗi lít dung dịch yêu cầu. Đun sôi hỗn hợp này trong 1 - 2 phút và rồi rót vào nước lạnh. Làm đầy đến thể tích yêu cầu trong bình hút, lắc, và bảo quản dung dịch này trong cacbon dioxyt (CO2).
Phủ lên dung dịch một lớp paraffin lỏng (dày khoảng 0,6 cm).
Chuẩn hoá dung dịch titan clorua: Cân 3 g amoni sắt sunfat [(NH4)2 SO4 . FeSO4 . 6H2O] cho vào một bình nón 500 ml và dẫn một luồng khí cacbon dioxit CO2 qua bình nón liên tục cho đến khi kết thúc xác định. Thêm 50 ml nước và 25 ml dung dịch axit sunfuric 10 N rồi thêm 30 ml dung dịch kali dicromat 0,1 N, đã được chuẩn hoá chính xác. Chuẩn độ với dung dịch titan clorua đến khi điểm kết thúc theo tính toán gần đạt tới. Thêm 5 ml dung dịch amoni thioxyanat 20 % và tiếp tục chuẩn độ đến khi màu đỏ mất đi và dung dịch giữ màu xanh. Tiến hành xác định mẫu trắng với 3 g amoni sắt sunfat dùng cùng một lượng nước, axit và dung dịch amoni thioxyanat và cũng cho dòng CO2 liên tục qua bình như trên.
Hệ số cho dung dịch titan clorua 0,1 N là:
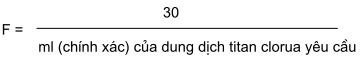
3.3.7.3 Xác định tổng hàm lượng chất màu của mẫu
Cân chính xác một lượng mẫu chất màu, được qui định theo từng chuyên luận, cho vào một bình thuỷ tinh dung tích 500 ml và thêm 10 g natri xitrat hay 15 g natri hydrotactrat, qui định theo từng chuyên luận, và thêm 150 ml nước. Cho một dòng khí CO2 đi qua bình, đun nóng dung dịch đến sôi, và chuẩn độ với dung dịch titan clorua đã chuẩn hoá giữ dòng CO2 ổn định. Gần đến điểm kết thúc chuẩn độ, thêm dung dịch titan clorua từng giọt đến điểm kết thúc. Chất màu giữ vai trò làm chỉ thị của chính bản thân nó, trừ khi có chỉ dẫn trong chuyên luận tương ứng.
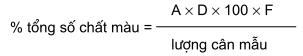
trong đó
A là số mililit dung dịch titan clorua 0,1 N cần dùng (chính xác);
D là lượng chất màu tương đương với 1,00 ml dung dịch titan clorua 0,1 N (được xác định trong mỗi
tài liệu).
3.3.7.4 Qui trình xác định phẩm màu đỏ tía (lakes)
Cho 150 ml nước vào bình nón dung tích 500 ml, hoà tan trong đó chất đệm được chỉ định cho chất màu gốc. Cân chính xác một lượng phẩm màu đỏ tương đương với 30 ml - 40 ml dung dịch titan clorua 0,1 N và chuyển vào một bình nón. Đun đến sôi hoặc đến khi phẩm màu đỏ tía tan hoàn toàn. Chuẩn độ với dung dịch titan clorua như cách chỉ dẫn ở trên.
3.3.8 Phương pháp thử các phẩm màu thực phẩm nhất định (dự kiến)
3.3.8.1 Cách tiến hành
Hoà tan một lượng chất màu tương đương với giá trị (p) trong bảng sau, vào amoni axetat (1,542 g trong một lít nước); pha loãng 1 ml dung dịch này đến 100 ml với cùng dung dịch amoni axetat trên.
Trong cùng một điều kiện như nhau, chuẩn bị một dung dịch thử với (p') mg chất màu so sánh.
Xác định bằng phép đo phổ nhìn thấy được tại bước sóng của chất màu so sánh ± 5 nm, độ hấp thụ của dung dịch thử (A1) và của dung dịch so sánh (A2).
3.3.8.2 Tính toán hàm lượng phần trăm chất màu trong mẫu kiểm tra như sau:

trong đó
T là nồng độ (%) của chất màu tinh khiết trong chất màu so sánh.
Bảng 2
|
|
p, p' |
λ nm |
C.l |
|
Azorubine Brillant xanh FCF Patent xanh V Ponceau IV R Erythrosine Indigotine Sunset vàng Quinolin vàng Brillant đen Tartrazine |
160 mg 40 mg 50 mg 170 mg 75 mg 150 mg 150 mg 100 mg 150 150 |
561 nm 630 nm 640 nm 507 nm 525 nm 612 nm 480 nm 416 nm 573 426 |
14720 42900 42051 16255 45430 73015 15985 47005 28840 19140 |
3.3.9 Hàm lượng chất màu phụ
3.3.9.1 Lưu ý chung
Trong niều năm sắc ký giấy đã được sử dụng cho việc xác định hàm lượng các chất mầu phụ của phẩm màu thực phẩm tan trong nước. Kĩ thuật đã được xây dựng và mô tả để được sử dụng thông dụng và được thừa nhận rằng sự hấp thụ của các chất màu phụ cũng giống như các chất mầu chính. Theo đó các chất chuẩn của từng chất mầu phụ thì không cần đến.
Kỹ thuật HPLC (sắc ký lỏng - High - performance liquid chromatography) đã được sử dụng thành công để tách chiết và xác định hàm lượng các chất mầu phụ của một số phẩm màu thực phẩm, gồm cả một số phẩm màu tan trong nước, các chất chuẩn của từng chất mầu phụ riêng biệt là cần thiết cho phương pháp này. Mặc dầu vậy phải nhớ rằng, đặc tính các dạng giới hạn, … trừ phi có công bố khác thì có liên quan đến phương pháp sắc ký giấy và các điều kiện được tra cưú ở phần "phép thử".
3.3.9.2 Định nghĩa
Các chất mầu phụ là các chất mầu được tạo ra trong quá trình sản xuất để bổ xung cho chất mầu được đặt tên chính. Bất kỳ một chất màu nào khác ngoài chất mầu chính và phụ đều được coi như giả mạo và thông thường có thể phát hiện được chúng trên sắc đồ dùng trong quá trình xác định các chất mầu phụ. Việc đọc (suy luận) trên sắc đồ tìm các chất mầu giả thường đòi hỏi ít nhiều kinh nghiệm.
3.3.10 Xác định bằng phương pháp sắc ký giấy
3.3.10.1 Nguyên tắc
Các chất mầu phụ được tách ra từ chất mầu chính bằng cách cho chạy sắc ký ngược trên giấy và được chết tách riêng rẽ trên giấy sắc ký. Độ hấp thụ của mỗi chất chiết được đo ở bước sóng hấp thụ tối đa của nó trong vùng phổ nhìn thấy được.
Vì việc nhận biết từng chất mầu phụ trong mỗi phẩm mầu thực phẩm là không thực tiễn, nên người ta chấp nhận một phương pháp gần đúng, thể hiện chúng dưới dạng phần trăm của mẫu thử. Ta thừa nhận khả năng hấp thụ của từng chất màu phụ là giống với khả năng hấp thụ của tổng các chất mầu.
Độ hấp thụ của các chất chiết được gộp chùng vào, và sử dụng cùng với độ hấp thụ của mẫu thử và hàm lượng của tổng các chất mầu để tính toán hàm lượng các chất mầu phụ. Điều này được coi là một phép tính xấp xỉ gần đủ cho việc xác định một thành phần thứ yếu.
3.3.10.2 Thiết bị:
- Bình sắc ký và thiết bị phụ trợ.
Hình 1 và 2 cho thấy thiết bị phù hợp gồm có:
• bình thuỷ tinh (A) và nắp đậy (B);
• khung đỡ (C) cho các tờ giấy sắc ký;
• khay (D) cho dung môi khai triển;
• khung thứ 2 (E) để đỡ các "tấm mành" giấy lọc;
• các tờ giấy sắc ký, kích thước không nhỏ hơn 20 cm x 20 cm.
(Giấy sắc ký Whatman No.1 là phù hợp).
- Micrô xơ - ranh, dung tích phân phối 0,1 ml, có sai số chấp nhận là ± 0,002 ml.
- Máy đo quang phổ.
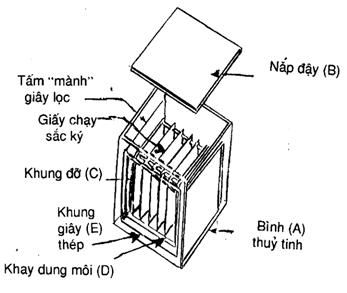
Hình 2 - Bộ phận của thiết bị sắc ký
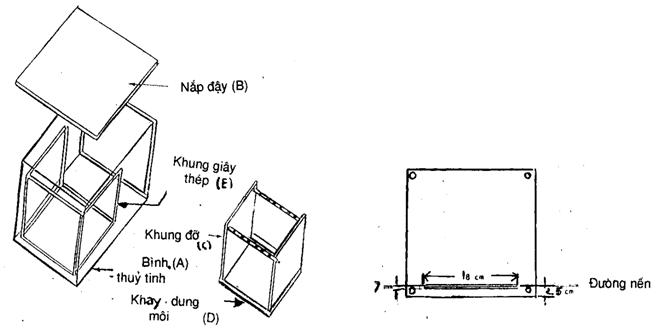
Hình 3 - Các chi tiết của thiết bị sắc ký Hình 4 - Phương pháp vẽ lên giấy sắc ký
3.3.10.3 Cách tiến hành
ít nhất 2 giờ (h) trước khi tiến hành phép thử, đặt một tấm giấy lọc vào bình khai triển; rót lên các lớp giấy này và rót vào đáy bình khai triển một lượng dung môi đủ để phủ lên đáy bình một lớp dày khoảng 1 cm. Đặt khay dung môi (D) vào vị trí và đậy nắp bình lại.
Đánh dấu lên 1 tờ giấy sắc kí như trong hình 3. Chấm 0,10 ml dung dịch mẫu 1,0 % trong nước, càng xơ- ranh đều càng tốt trong phạm vi giới hạn là 1 hình chữ nhật kích thước 18 x 7 mm, giữ chắc chắn đầu micro xơ ranh (xơ - ranh vi lượng) khi chạm vào giấy.
Để cho giấy này khô ở nhiệt độ phòng trong 1 h - 2 h, hoặc ở 500C trong 5 phút, sau đó thêm 15 phút ở nhiệt độ phòng. Gắn tờ sắc ký này, cùng với 1 tờ giấy sắc ký không chấm để làm màu trắng vào khung (C).
Rót một lượng vừa đủ dung môi sắc ký vào khay (D), sao cho bề mặt lớp dung môi này thấp hơn đường nền của tấm giấy sắc ký khoảng 1 cm. Thể tích dung môi cần dùng sẽ tuỳ thuộc vào kích thước bình khia triển và phải được xác định trước. Đặt khung (C) vào vị trí và đậy lại nắp. Để cho dung môi chạy lên phía trước đến khoảng cách đã định phía trên đường nền, sau đó nhấc khung (C) ra và đưa nó vào buồng sấy khô ở 500 - 600 trong 10 - 15 phút. Lấy giấy đã chạy sắc ký ra khỏi khung (C).
(Có thể cho khai triển một vài sắc tố đồ đồng thời, nếu thấy cần thiết).
Cắt từ tấm giấy sắc kí ra thành từng dải băng màu phụ, và cắt lấy một dải giấy tương đương từ vị trí tương ứng trên tấm giấy đối chứng. Đặt mỗi dải, đã chia thành một số phần phù hợp, xấp xỉ nhau, đặt vào một ống nghiệm riêng. Thêm 5,0 ml hỗn hợp nước: axeton (1 : 1 theo thể tích) vào mỗi ống nghiệm, lắc tròn trong 2 - 3 phút thêm 15,0 ml dung dịch natri hidro cacbonat 0,05 N và lắc ống nghiệm để trộn đều. Lọc các dịch chiết có màu và mẫu trắng qua giấy lọc đường kính 9 cm loại đính tính (giấy thưa) và xác định độ hấp phụ của dịch chiết màu này ở bước sóng hấp thụ tối đa của chúng, sử dụng cuvet 40 mm đậy kín, so sánh với hỗn hợp đã lọc gồm 5,0 ml nước: axeton (1 : 1 theo thể tích) và 15,0 ml dung dịch natri hidro cacbonat 0,05 N. Đo độ hấp thụ của dịch chiết từ dải mầu trắng ở cùng bước sóng đã đo dịch chiết màu và hiệu chính độ hấp thụ của các dịch chiết màu với các giá trị đo mẫu trắng.
Để chuẩn bị vết "chuẩn", tiến hành như sau:
Từ dung dịch 1 % mẫu thử, chuẩn bị một dung dịch tương ứng với L/100%, trong đó L là giới hạn các chất màu phụ. Chấm 0,10 ml dung dịch này lên một tấm giấy sắc kí bằng kĩ thuật đã mô tả ở trên, cho chạy sắc đồ, sau đó làm khô nó ở 500 - 600 trong 10 - 15 phút. Cắt băng giấy từ tấm giấy này thành 1 dải và cắt một dải tương tự từ một tấm giấy trắng nhưng có đánh dấu. Tiến hành các bước như trên và xác định độ hấp phụ thực (As) của chất chuẩn.
3.3.10.4 Tính toán phần trăm các chất màu phụ theo:
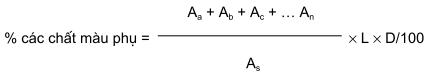
trong đó
Aa + Ab + Ac + … An là tổng độ hấp thụ của các chất màu phụ được hiệu chỉnh theo giá trị mẫu trắng;
D là tổng hàm tổng các chất của mẫu thử.
3.3.10.5 Các dung môi chạy sắc kí:
1. Nước: amoniac (tỉ trọng 0,880): trinatrixitrat (95 ml : 5 ml : 2 g)
2. n-Butanol: nước: etanol: amoniac (tỉ trọng 0,880) (600 : 264 : 135 : 6)
3. Butan - 2 - one: axeton: nước (7 : 3 : 3)
4. Butan - 2 - one: axeton: nước: amoniac (tỉ trọng 0,880) (700 : 300 : 300 : 2)
5. Butan - 2 - one: axeton: nước: amoniac (tỉ trọng 0,880) (700 : 160 : 300 : 2)
6. n- Butanol: axit axetic băng: nước (4 : 1 : 5)
Lắc trong 2 phút để yên cho các lớp tách ra. Sử dụng lớp trên bên trên làm dung môi sắc kí.
3.3.11 Phép thử nhận biết và giới hạn nhanh bằng sắc kí lớp mỏng (dự kiến)*)
3.3.11.1 Nguyên tắc
Các tạp chất màu được xác định bằng sắc kí lớp mỏng (T. L. C) trên một bản mỏng silicagen.
3.3.11.2 Chuẩn bị các dung dịch:
Dung dịch 1: Hoà tan 40 mg chất màu thực phẩm cần thử trong hỗn hợp metanol/ nước (50/50) và pha loãng đến 10 ml bằng chính dung môi đó.
Dung dịch 2: Pha loãng 0,1 - 0,5 ml **) dung dịch 1 thành 10 ml bằng hỗn hợp metanol/ nước (50/50).
Dung dịch 3 (dung dịch chuẩn): Hoà tan 40 mg phẩm màu chuẩn tương ứng trong hỗn hợp metanol/nước (50/50) và pha loãng đến 10 ml bằng chính dung môi đó.
3.3.11.3 Cách tiến hành
Chấm 5 μl từ dung dịch 1; dung dịch 2 và dung dịch chuẩn. Đặt bản mỏng vào buông khai triển có chứa n- butanol/ etanol/ nước/ dung dịch amoniac (50 / 25 / 25 / 10) và để cho dung môi chạy tới 1 điểm ở phía trên cách vết chấm mẫu là 15 cm.
Các vết chấm chính xuất hiện trên sắc đồ 1 và 3 dịch chuyển gần tới cùng giá trị Rf. Bất kỳ vết nào khác xuất hiện trên sắc đồ 1 thì sẽ không lớn hơn vết chính xuất hiện trên sắc đồ 2.
Chú thích - Đôi khi có thể rửa (tẩy) các vết chấm trên sắc đồ 1 và thực hiện một phép thử gần đúng. Trong trường hợp này, cách tiến hành được mô tả ở tài liệu FNP 31/ 1 (1984), trang A. 10 có thể được cải tiến như sau:
"Cạo từng điểm phụ ra khỏi bản mỏng và cạo điểm tương đương ở vị trí tương ứng trên bản trắng. Đặt từng…"
3.4 Chất có thể chiết được bằng ête
3.4.1 Phương pháp I
3.4.1.1 Thiết bị
*) Việc sử dụng phương pháp này có thể thay đổi.
**) Tuỳ theo giới hạn của các tạp chất màu hoặc các màu phụ được phép có trong chất màu kiểm tra (1 đến 5 %)
Bộ chiết chất lỏng / chất lỏng kiểu hướng lên trên, có bộ phận phân phối bằng thuỷ tinh xốp, dung tích làm việc là 200 ml. Một mẩu dây đồng thau được treo lơ lửng qua bộ ngưng và một miếng dây đồng nhỏ, tròn (0,5 g) được cho vào bình cất.
3.4.1.2 Ête : Dietyl ête hoặc diisopropyl ête
Ngay trước khi sử dụng, cần phải cho ête mới chưng cất đi qua một cột oxit nhôm - loại dành cho sắc kí - cao 30 cm, để loại bỏ các peroxit và các chất cản. Thử để đảm bảo là đã hết peroxit theo cách sau:
Pha một dung dịch sắt thioxyanat không màu bằng cách trộn một lượng dung dịch sắt sunfat 0,1 N với một lượng tương đương amoni thioxyanat và loại bỏ hết bất kì vết màu đỏ nào - do ion sắt bằng titan clorua. Thêm 10 ml ête vào 50 ml dung dịch này và lắc hỗn hợp thật mạnh trong 2 - 3 phút. Không có màu đỏ xuất hiện nữa.
3.4.1.3 Cách tiến hành
a) Chiết ête tính kiềm: cân chính xác khoảng 5,0 g mẫu phẩm màu, hoà tan trong 150 ml nước, thêm 2,5 ml dung dịch NaOH 2 N và chuyển dung dịch này vào dụng cụ chiết, pha loãng bằng nước tới 200 ml. Thêm 200 ml ête vào bình cất và chiết trong hai giờ với tốc độ hồi lưu khoảng 15 ml/phút. Giữ lại dung dịch màu. Chuyển dịch chiết ête này vào một phễu chiết và rửa dịch chiết ête với 2 lượng (lần) ì 25 ml NaOH 0,1 N và sau đó rửa bằng nước. Cho chưng cất ête từng phần từ một bình dung tích 150 ml, đã cân khối lượng trước, có chứa một miếng đồng tròn, sạch, cho giảm thể tích tới khoảng 5 ml.
b) Chiết ête tính axit: thêm vào dung dịch màu được giữ lại từ (a) - 5 ml axit HCl 3 N, trộn và đem chiết một lượng ête nữa như trong mục (a). Rửa dịch chiết ête bằng 2 lần x 25 ml axit HCl 0,1 N và sau đó rửa bằng nước. Chuyển từng phần vào bình chứa chất chiết kiềm tính đã bốc hơi, và cẩn thận làm bay hơi toàn bộ ête. Sấy khô hoàn toàn trong tủ sấy ở 850 trong 20 phút, sau đó để cho bình thí nghiệm này nguội đi trong bình hút ẩm 30 phút, và đem cân. Lặp lại quá trình sấy khô và làm nguội cho đến khi có khối lượng không đổi. Độ tăng về khối lượng của bình thử nghiệm đã biết trước khối lượng, được biểu diễn bằng phần trăm khối lượng của mẫu thử, chính là "chất có thể chiết được bằng ête".
3.4.2 Phương pháp ll
3.4.2.1 Thiết bị: Bộ chiết soclet. Một mẩu dây đồng thau được treo lơ lửng qua phần ngưng và một dây đồng nhỏ (0,5 g) được cho vào bình chưng cất.
3.4.2.2 Ête: Dietyl ête hoặc diisopropyl ête
Tinh chế ête như chỉ dẫn trong phương pháp l.
3.4.2.3 Cách tiến hành
Cân chính xác khoảng 2 g mẫu phẩm màu. Chuyển vào ống chiết soclet (thimble) và chiết với 150 ml ête trong 5 giờ. Đem cô đặc dịch chiết ête trên thiết bị cách hơi nước còn khoảng 5 ml. Làm khô phần cặn này trong một "đĩa bốc hơi" đã biết khối lượng, trong một bếp cách thuỷ, sau đó sấy khô ở 1050 đến khi có khối lượng không đổi.
Độ tăng về khối lượng ở "đĩa bay hơi", được biểu thị bằng phần trăm khối lượng của mẫu thử, chính là "chất có thể chiết bằng ête".
3.5 Các chất có trong phẩm màu đỏ tía (lakes) không tan trong axit clohydric
3.5.1 Thuốc thử
- Axit clohydric đậm đặc;
- Axit clohydric nồng độ 0,5 % V/V.
3.5.2 Cách tiến hành
Cân chính xác khoảng 5 g phẩm màu đỏ tía cho vào một cốc có mỏ dung tích 500 ml. Thêm 200 ml nước và 60 ml axit clohydric đậm đặc. Đun sôi đến khi hoà tan toàn bộ màu và alumin. Lọc qua phễu lọc xốp số 4 đã biết khối lượng. Rửa phễu lọc này bằng axit clohydric HCl 0,5 % nóng cho đến khi nước rửa không còn màu. Sấy khô phễu đến khối lượng không đổi ở 1350C. Biểu thị khối lượng của cặn bằng phần trăm khối lượng đã lấy.
3.6 Nền trắng trong các chất màu triarylmetan được sunfonat hoá
3.6.1 Nguyên tắc
Không khí được thổi qua một dung dịch nước có chứa clorua và dimetylformamit. Trong điều kiện này, chất nền trắng được oxi hoá thành các chất màu và độ tăng khả năng hấp thụ là số đo lượng chất nền trắng vốn có (trong mẫu).
3.6.2 Thuốc thử
* Dimetylformamit (DMF)
- Dung dịch A: Cân 10,0 g CuCl2 . 2H2O và hoà tan trong 200 ml DMF. Chuyển vào một bình định mức dung tích 1 L và thêm DMF làm đầy đến vạch mức.
- Dung dịch B: Cân chính xác khối lượng mẫu thử đã định. Hoà tan trong khoảng 100 ml nước, chuyển lượng đó vào một bình định mức dung tích 1 L và thêm nước tới vạch mức.
3.6.3 Cách tiến hành
Chuẩn bị các dung dịch sau:
- Dung dịch a: Dùng pipet cho 50 ml DMF dung dịch A vào một bình định mức dung tích 250 ml.
Đậy bằng màng "parafilm" và đặt vào chỗ tối.
- Dung dịch b: Dùng pipet cho chính xác 10 ml dung dịch B vào một bình định mức dung tích 250ml. Thêm 50 ml DMF. Đậy bằng màng "parafilm" và đặt vào chỗ tối.
- Dung dịch c: Dùng pipet hút 50 ml DMF vào một bình định mức dung tích 250 ml. Cho sục khí qua dung dịch này trong 30 phút theo cách dưới đây:
Luồn một pipet 5 ml vào một hộp có gắn với một nguồn dạng uốn cong để thổi không khí. Cho khí thổi qua một cách từ từ. Nhưng đầu pipet vào dung dịch chứa trong bình và điều chỉnh dòng khí với một tốc độ nhanh nhưng kiểm soát được. Sau 30 phút, kéo pipet trên ra khỏi dung dịch và tráng, rửa các thành pipet bằng nước, cho vào trong bình, sử dụng bình tia. Sau đó, tắt dòng không khí.
- Các dung dịch d1 và d2:
Dùng pipet hút chính xác 10 ml dung dịch B cho vào hai bình định mức riêng rẽ, dung tích 250 ml, theo cùng cách như đã làm với dung dịch b. Thêm 50 ml dung dịch A vào mỗi bình. Cho sục không khí qua các dung dịch này trong 30 phút, sử dụng phương pháp nêu trên.
Sau khi đã sục không khí nhanh qua các dung dịch trên, cho thêm nước vào tất cả 5 bình này gần tới vạch mức. Khi DMF và nước trộn với nhau sẽ có phát nhiệt, vì vậy đặt các bình trên vào một thùng nước (nước lấy từ vòi nước) cho đến khi chúng nguội xuống nhiệt độ phòng. Đừng giữ các bình lâu hơn mức cần thiết; thông thường 5 - 10 phút là đủ. Đổ thêm nước chính xác đến vạch mức. Đo ngay các dung dịch này trên máy quang phổ. Toàn bộ tiến trình được thực hiện càng nhanh càng tốt.
3.6.4 Xác định quang phổ
Vẽ các đường cong dưới đây từ bước sóng 700 - 500 nm, sử dụng một dải hấp thụ các cuvet 0,1 và 1 cm. Cho chạy tất cả các đường cong trên cùng một đồ thị phổ, và (để chính xác tối đa) bỏ đi các số đọc trên màn hình hiện số ở bước sóng tối đa giữa 620 nm và 635 nm bằng cách quay ngược lại sau khi đường cong được dựng lên.
3.6.5 Mẫu so sánh
|
Đường cong |
Cuvet |
Cuvet |
Nhận xét |
|
1 |
a |
a |
Đặt điểm 0 ở 700 nm, cho chạy; ghi lại độ hấp thụ ở Abs chuẩn đối với chất màu |
|
2 |
a |
b |
Cho chạy đường cong mà không đặt điểm 0 hiệu chỉnh. Ghi lại độ hấp thụ tối đa. |
|
3 |
c |
c |
Đặt điểm 0 ở 700 nm; ghi độ hấp thụ Abs chuẩn đối với chất màu. |
|
4a |
c |
d1 |
Chạy đường cong không đặt điểm 0 hiệu chỉnh.Ghi lại độ hấp thụ tối đa. |
|
4b |
c |
d2 |
Chạy đường cong không đặt điểm 0 hiệu chỉnh;ghi độ hấp thụ tối đa. (d1 và d2 là các lần đo song hành) |
Chú thích - Các cu vét cần phải tráng rửa kỹ trước mỗi lần đo. Để rửa sạch theo dọc cuvet, sử dụng 3 lần tráng riêng rẽ với ít nhất 40 ml dung dịch mẫu thử.
3.6.6 Tính toán
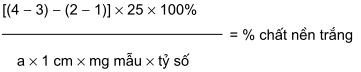
trong đó
a là khả năng hấp thụ 100% các chất màu;
mg mẫu là lượng mẫu thử được cân, tính bằng mg;
|
tỷ số = |
MW của chất màu |
(được cho trong các tài liệu riêng); |
|
MW của chất nền trắng |
1, 2, 3 và 4 là độ hấp thụ tối đa của các cuvet 1, 2, 3 và 4.
3.7 Các hợp chất hữu cơ khác ngoài chất màu
3.7.1 Chú thích chung
Để tách và xác định các hợp chất không màu, sắc kí lỏng (HPLC) có một vài ưu thế hơn các kỹ thuật sắc kí khác, đó là: cải thiện được quá trình tách, tốc độ, (có thể tự động hoá), và độ chính xác. Khi xác định tên các hợp chất hữu cơ, sẽ cần đến một mẫu thử từ mỗi nguyên liệu có thể bắt gặp, trước khi phân tích bất kì một màu cụ thể nào (đặc biệt). Thông thường phương pháp HPLC được phác thảo hơn là mô tả chi tiết.
Nên nhớ rằng sắc kí lỏng vẫn đang ở thời kì phát triển đáng kể. Các nguyên liệu nhồi cột, các cột mao dẫn, kiểu và độ nhậy của các detectoz là một vài khía cạnh vẫn đang tiếp tục thu hút sự chú ý của các nhà sản xuất và do thế, sự phát triển chúng có thể dẫn đến việc tách được các tạp chất, ngoài những chất thường xuyên được kể đến trong bản mô tả một phẩm màu thực phẩm nào đó.
Phương pháp được chọn sử dụng một cột trao đổi anion, nhưng các cột pha ngược đã dùng một cách rộng rãi và cho độ tách lý tưởng.
Phương pháp khác (truyền thống) dùng sắc kí cột, kĩ thuật này nhằm thu thập các phần rửa giải phân đoạn, và sử dụng phổ hấp thụ tử ngoại của chúng để nhận biết các hợp chất có mặt, và để tính toán nồng độ các chất này.
3.7.2 Xác định bằng sắc kí lỏng
Các hợp chất hữu cơ - ngoài chất màu - được tách ra bằng sắc kí lỏng, có sử dụng gradien rửa giải và được định lượng bằng cách so sánh khoảng "pic" của chúng với các "pic" thu được từ các (dung dịch) chuẩn đã biết. Các điều kiện mô tả ở đây cần xử lý như những hướng dẫn mà thôi, và cũng cần đến các sửa đổi thêm để đạt được độ tách tốt.
Các điều kiện để tiến hành sắc kí được cho dưới đây:
1) Thiết bị (Instrument) - sắc kí lỏng cao áp, có lắp một bộ phận phù trợ: gradien rửa giải.
2) Detector - Detector UV (tử ngoại) ghi độ hấp thụ ở bước sóng 254 nm.
3) Cột - bằng thép không rỉ, 1 m x 2,1 mm (đường kính trong).
4) Chất nhồi cột - chất trao đổi anion mạnh màng mỏng (SAX). Ví dụ: một phần t- amon được thay thế lớp phủ polime metacrylat 1 % theo khối lượng.
5) Nồng độ mẫu thử: - 2 % (khối lượng / khối lượng) trong dung dịch natri tetraborat 0,01 M.
6) Thể tích bơm - 10 àl.
7) Hệ dung môi –
• thứ nhất: natri tetraborat 0,01 M.
• thứ hai: natri tetraborat 0,01 M.
natri peclorat 0,1 M
8) Gradient - xem theo từng chuyên luận.
9) Tốc độ dòng - 1,0 ml/phút.
10) Nhiệt độ - phòng.
Các điều kiện thực nghiệm khác như: chiều dài cột, loại chất nhồi cột và hệ dung môi, và cách thức sử dụng các cặp ion có thể cho các sai lệch (biến thiên) về đặc tính rửa giải như trật tự và độ chuyển đổi.
3.7.3 Xác định bằng sắc kí cột
3.7.3.1 Thiết bị
(ống) Cột sắc kí (xem hình dưới). Máy đo quang phổ phù hợp cho sử dụng ở vùng ngoại.
Đơn vị: cm
3.7.3.2 Chuẩn bị cột sắc kí
Chuẩn bị một khối bột xenluloza nhuyễn loại Whatma, trong dung dịch amoni sunfat 25%. Nếu dùng loại xenluloza khác, cần phải là loại có hàm lượng sắt (Fe) rất thấp.
Thử nghiệm: Chuẩn bị cột như chỉ dẫn và cho 200 ml dung dịch amoni sunfat 25% chảy qua cột. Độ hấp thụ tử ngoại của dung dịch này phải đủ thấp để tránh gây nhiễu cho chất định phân tích. Dùng khoảng 75 g xenluloza cho 500 ml chất lỏng. Đặt một đĩa nhỏ vào lưới thép không rỉ vào chỗ thắt phía trên vòi phễu của ống sắc kí. Rót một lượng vừa đủ bột nhào vào ống (sắc kí) để có một cột với độ cao cách miệng ống khoảng 5 cm. Thỉnh thoảng vỗ nhẹ vào ống để đảm bảo cột được nhồi chặt. Rửa cột bằng 200 ml chất rửa trôi.
3.7.3.3 Cách tiến hành
Cho 0,200 g mẫu phẩm màu vào một cốc có mỏ thích hợp, và hoà tan trong 20 ml nước. Thêm khoảng 5 g bột xenluloza. Thêm 50 g amoni sunfat để muối hoá phẩm màu. Chuyển hỗn hợp này vào ống sắc kí, tráng cốc bằng dung dịch amoni sunfat 25% và cho nước tráng này vào ống sắc kí. Để cho cột rút nước cho đến khi ngừng chảy, hoặc gần như ngừng chảy.
Thêm dung dịch amoni sunfat vào cột ở tốc độ tương đương với tốc độ dòng chảy qua cột. Thu vào mỗi ống 100 ml dung dịch chảy ra theo từng phân đoạn. Tiếp tục cho đến khi thu được 12 phần tách như trên. Giữ lại cột và các chất trong cột cho đến khi đã kiểm tra xong phần dịch tách cuối cùng.
Lắc trộn kĩ từng phần dịch tách được, và đo phổ hấp thụ tử ngoại của mỗi dung dịch bước sóng từ 220 nm - 400 nm. Nếu phổ hấp thụ của phần dịch thứ 12 cho thấy sự có mặt của bất kỳ một hợp chất nào, thì tiếp tục thu các phần tách ra từ cột cho đến khi các hợp chất có trong đó đã được rửa trôi hết.
Thường thường chỉ gặp một hợp chất. Việc nhận biết và định lượng được hoàn tất bằng cách so sánh phổ hấp thụ của chất được rửa trôi với phổ hấp thụ của dung dịch hợp chất tinh khiết này trong cùng loại dung môi.
Khi có mặt nhiều hơn một hợp chất, với một lượng đáng kể trong bất kì phần dịch tách phân đoạn nào, các số liệu đo quang phổ sẽ chỉ cho thấy điều đó. Trong trường hợp này, khối lượng các hợp chất khác nhau đó cần phải xác định quá trình sử dụng trong phân tích quang phổ cuả các hỗn hợp các chất hấp thụ.
Một vài mẫu thử có chứa lượng nhỏ các chất khác nhau, đặc biệt là các muối vô cơ, sẽ cho "độ hấp thụ nền". Hiệu chỉnh điều này như sau:
Xác định lượng chất hấp thụ này ở phần dịch thu từ cột ngay trước, và liền sau các phần dịch tách phân đoạn mà ta bắt gặp các hợp chất trên. Lấy độ hấp thụ quan sát được của các phần dịch tách có chứa hợp chất đó từ đi một nửa (1/2) của tổng hai lần xác định này. Phần còn lại (hiệu) được lấy làm độ hấp thụ do có hợp chất trên.
3.8 Gốc sunfat, tính theo natri sunfat
3.8.1 Cân 5,0 g mẫu phẩm màu, cho vào một bình nón dung tích 250 ml và hoà tan trong khoảng 100 ml nước bằng cách hâm nóng trong bình cách thuỷ. Thêm 35 g natri clorua không chứa sunfat. Đậy nút bình, và lắc tròn định kì trong vòng 1 giờ. Làm nguội, chuyển bằng dung dịch natri clorua bão hoà, vào một bình định mức dung tích 250 ml, và pha loãng tới vạch mức ở 200. Lắc bình, và đem lọc dung dịch này qua một giấy lọc khô. Dùng pipet hút 100 ml dịch lọc cho vào một cốc thuỷ tinh 500 ml, pha loãng tới 300 ml bằng nước và axit hoá bằng axit clohydric, thêm 1 ml dư. Đun dung dịch tới sôi, và thêm một lượng dư dung dịch bari clorua 0,25 N, từng giọt, vừa thêm vừa khuấy. Để yên hỗn hợp này trên bếp điện trong 4 h, hoặc để qua đêm ở nhiệt độ phòng và sau đó gia nhiệt tới khoảng 800 và để cho tủa lắng xuống. Lọc bỏ tủa bari sunfat, rửa bằng nước nóng và đốt ở độ nóng màu đỏ đậm, trong một chén nung đã biết khối lượng, cho đến khi thu được khối lượng không đổi.
3.8.1 Tiến hành xác định một mẫu trắng, áp dụng hiệu chỉnh cần thiết về khối lượng bari sunfat tìm thấy trong phép thử (nếu có), và tính toán kết quả theo natri sunfat.
Khối lượng natri sunfat trong mẫu thử = 2,5 x khối lượng đã hiệu chỉnh của bari sunfat x 0,6086
Trình bày kết quả dưới dạng phần trăm khối lượng mẫu thử đã lấy.
3.9 Các gốc amin thơm không sunfonat hoá
3.9.1 Nguyên tắc
Các gốc amin thơm không sunfonat hoá được chiết tách trong toluen từ một dung dịch mẫu thử kiềm tính, được chiết lại trong axit và sau đó đem đo trên máy quang phổ, sau khi đã diazo hoá 2 lần và kết hợp cả 2 lần.
Chúng được trình bày dưới dạng anilin, trừ khi đã biết trước chúng là dạng amin khác nào đó.
3.9.2 Thiết bị
Máy đo quang phổ đo ở dải nhìn thấy được.
3.9.3 Thuốc thử
- Các thuốc thử phải được công nhận có chất lượng thuốc thử phân tích.
- Nước cất, hoặc ít nhất là nước có độ tinh khiết tương đương.
1. Toluen;
2. Axit clohydric, dung dịch xấp xỉ 1 N;
3. Axit clohydric, dung dịch xấp xỉ 3 N;
4. Kali bromua, dung dịch xấp xỉ 50 %;
5. Natri cacbonat, dung dịch xấp xỉ 2 N;
6. Natri hydroxit, dung dịch xấp xỉ 1 N;
7. Natri hydroxit, dung dịch xấp xỉ 0,1 N;
8. Muối R (muối 2 - naphtol - 3,6 axit disunphonic, dinatri), dung dịch xấp xỉ 0,05 N;
9. Natri nitrit, dung dịch xấp xỉ 0,5 N;
10. Dung dịch anilin chuẩn.
Cân 0,100 g anilin đã cất lại vào một chén cân nhỏ, sau đó tráng rửa nó cho vào bình định mức, dung tích 100 ml, tráng cốc cân vài lần bằng nước. Thêm 30 ml dung dịch axit clohydric xấp xỉ 3 N và pha nước tới vạch, ở nhiệt độ phòng. Gọi là dung dịch A.
Đem pha loãng 10,0 g dung dịch A bằng nước tới 100 ml trong bình định mức, và trộn kĩ. Gọi là dung dịch B; 1 ml dung dịch này tương đương với 0,0001 g anilin. (Khi sử dụng mới pha dung dịch B).
3.9.4 Cách tiến hành
3.9.4.1 Chuẩn bị đồ thị chuẩn
Đong vào một dãy các bình định mức, dung tích 100 ml, lần lượt các thể tích anilin chuẩn, (dung dịch B) như sau: 5 ml, 10 ml, 15 ml, 20 ml, 25 ml.
Pha loãng tới 100 ml bằng dung dịch axit clohydric 1 N và trộn kĩ. Dùng pipet hút 10 ml từ mỗi hỗn hợp này cho vào các ống nghiệm khô, sạch và làm nguội trong 10 phút bằng cách ngâm vào một cốc có chứa hỗn hợp đá/nước. Thêm vào mỗi ống nghiệm 1 ml dung dịch kali bromua và 0,05 ml dung dịch natri nitrit. Trộn và để yên trong 10 phút trong bồn đá/nước. Lấy 5 bình định mức dung tích 25 ml, cho vào mỗi bình 1 ml dung dịch muối R và 10 ml dung dịch natri cacbonat. Rót mỗi dung dịch anilin đã xử lý vào một bình riêng có chứa dung dịch muối R và dung dịch natri cacbonat, tráng các ống nghiệm bằng một ít nước. Dùng nước pha loãng tới vạch mức. Đậy nút bình, trộn kĩ dung dịch trong bình và để yên 15 phút ở chỗ tối.
Đo độ hấp thụ của mỗi dung dịch kết hợp ở bước sóng 510 nm trong các cuvet 40 mm, sử dụng mẫu so sánh là một hỗn hợp gồm 10,0 ml dung dịch axit clohydric 1 N: 10,0 ml dung dịch natri cacbonat: 2,0 ml dung dịch muối R, pha loãng đến 25,0 ml nước. Dựng một số đồ thị tương quan giữa độ hấp thụ và khối lượng anilin trong mỗi phần 100 ml dung dịch anilin.
3.9.4.2 Chuẩn bị và kiểm tra dung dịch thử
Cân khoảng 2,0 g mẫu phẩm màu, chính xác đến 0,01 g. Cho vào 1 phễu chiết có chứa 100 ml nước.
Đổ theo thành phễu một lượng 50 ml nước nữa, lắc tròn để hoà tan mẫu thử, và thêm 5 ml dung dịch natri hydroxit 1 N. Chiết với 2 lượng ì 50 ml toluen và rửa dịch chiết toluen kết hợp bằng từng lượng 10 ml dung dịch natri hydroxit 0,1 N để loại hết các vết màu. Chiết phần toluen đã rửa này bằng 3 lần x 10ml dung dịch axit clohydric 3 N và pha loãng phần dịch chết gộp chung này tới 100 ml bằng nước. Trộn kĩ. Gọi là dung dịch T.
Dùng pipet hút 10,0 ml dung dịch T cho vào một ống nghiệm khô, sạch làm nguội trong 10 phút bằng cách ngâm trong một cốc có hỗn hợp đá/nước, thêm 1 ml dung dịch kali bromua và tiến hành tiếp như đã mô tả ở trên cho việc chuẩn bị đồ thị chuẩn, bắt đầu với việc thêm 0,05 ml dung dịch natri nitrit.
Sử dụng một dung dịch được chuẩn bị từ 10,0 ml dung dịch T, 10 ml dung dịch natri cacbonat và 2,0 ml dung dịch R, pha nước tới 25 ml, làm dung dịch so sánh trong phép đo độ hấp thụ.
Đọc trên đồ thị chuẩn khối lượng anilin tương ứng với độ hấp thụ quan sát được từ dung dịch mẫu thử.
3.9.5 Tính toán
Phần trăm các gốc amin thơm không sunfonat hoá (tính theo anilin) có trong mẫu thử.
|
= |
Khối lượng anilin x 100 |
|
Khối lượng mẫu thử đã lấy |
3.10 Hàm lượng nước (hay lượng hao hụt sau khi sấy khô)
3.10.1 Các chất phẩm màu có chứa các nhóm SO3Na hay COONa thường có tính hút ẩm, và bất kì lượng nước nào chúng giữ lại từ nơi sản xuất (hoặc sau đó hấp thụ từ khí quyển) nói chung sẽ hiện diện trong phẩm màu đó ở dạng hydrat. Khi những phẩm màu này được sấy khô ở 1350, độ hao về khối lượng có thể là tương đương với tổng hàm lượng nước, nhưng điều này không phải đúng cho mọi trường hợp. Ví dụ, Erytrosin và Ponceau 4 R, mỗi loại giữ (ngậm) 1 phân tử nước dạng kết tinh ở 1350, và thông thường trong thực tế phải lưu ý đến điều này khi tính tổng hàm lượng các chất chính có mặt trong mẫu thử.
3.10.2 Cách tiến hành
Cân 2,0 g - 3,0 g mẫu trong một chén cân đã biết khối lượng, có một nắp tròn. Chén cân thuộc dạng lùn, có đường kính khoảng 50 mm và cao 30 mm là thích hợp. Sấy nóng ở nhiệt độ đã mô tả ± 50 cho đến khi có được khối lượng không đổi. Biểu thị độ hao hụt khối lượng theo phần trăm của khối lượng mẫu đã lấy.
3.11 Chất không tan trong nước
Cân 4,5 - 5,5 g1) mẫu thử vào một cốc thuỷ tinh dung tích 250 ml. Thêm khoảng 200 ml nước nóng (800- 900), khuấy để hoà tan, và để cho dung dịch nguội tới nhiệt độ phòng. Lọc dung dịch này qua phễu lọc thuỷ tinh xốp G 4, đã biết khối lượng (B.S.1752, phễu lọc đĩa xốp được sử dụng trong phòng thử nghiệm) và rửa bằng nước lạnh cho đến khi dịch rửa không còn màu. Sấy khô phễu lọc và cặn ở 1350 cho đến khi có được khối lượng không đổi.
Biểu thị khối lượng của cặn này theo phần trăm khối lượng của mẫu thử đã lấy.
3.12 Các gốc clorua và sunfat tan trong nước có ở phẩm màu đỏ tía (lakes) có nguyên tố nhôm
Cân chính xác 10 g mẫu thử. Thêm 250 ml nước. Khuấy để mẫu thử hoà vào nước và sau đó khuấy định kì khuấy, trong vòng 30 phút. Đem lọc dung dịch.
Đong 50 ml dịch đã lọc, thêm 50 ml nước nữa và axit hoá bằng 5 ml dung dịch axit nitric 1,5 N. Xác định hàm lượng clorua bằng phương pháp chuẩn độ điện thế sử dụng cho các phẩm màu tan được.
Đong lấy 50 ml dịch lọc, pha loãng tới 300 ml bằng nước và axit hoá bằng axit clohydric, thêm 1 ml dư.
Đun dung dịch tới sôi và thêm một lượng dư dung dịch bari clorua 0,25 N, từng giọt, vừa thêm vừa khuấy. Thực hiện quá trình phân tích với công đoạn vô cơ hoá, lọc, và nung kết tủa như đã mô tả trong phương pháp dùng để xác định sunfat ở các phẩm màu tan được.
1) Chú thích - Một vài phẩm màu có tính tan ít hơn 5 g / 200 ml về việc sử dụng lượng cân ít hơn sẽ được trình bày trong chuyên luận, trong mục "kiểm tra"
