Nội dung toàn văn Tiêu chuẩn Việt Nam TCVN 7557-1:2005 (0060 Method – EPA) về Lò đốt chất thải rắn y tế - Xác định kim loại nặng trong khí thải - Phần 1: Quy định chung do Bộ Khoa học và Công nghệ ban hành
TIÊU CHUẨN VIỆT NAM
TCVN 7557-1 : 2005
LÒ ĐỐT CHẤT THẢI RẮN Y TẾ - XÁC ĐỊNH KIM LOẠI NẶNG TRONG KHÍ THẢI – PHẦN 1: QUY ĐỊNH CHUNG
Health care solid waste incinerators – Determination of heavy metals in fluegas – Part 1: General requirements
Lời nói đầu
TCVN 7557-1 : 2005 hoàn toàn tương đương với 0060 Method – EPA về kỹ thuật nhưng có thay đổi về biên tập.
TCVN 7557 – 1 : 2005 do Tiểu ban kỹ thuật Tiêu chuẩn TCVN/TC 146/ SC2 “ Lò đốt chất thải rắn y tế” biên soạn trên cơ sở dự thảo đề nghị của Viện Y học lao động và Vệ sinh môi trường – Bộ Y tế, Tổng cục Tiêu chuẩn Đo lường Chất lượng xét duyệt, Bộ Khoa học và Công nghệ ban hành.
TCVN 7557 gồm có các tiêu chuẩn sau, với tên chung Lò đốt chất thải rắn y tế - Xác định kim loại nặng trong khí thải.
TCVN 7557-1 : 2005, Phần 1: Quy định chung.
TCVN 7557-2 : 2005, Phần 2: Phương pháp xác định nồng độ thủy ngân bằng phương pháp quang phổ hấp thụ nguyên tử kỹ thuật hóa hơi lạnh.
TCVN 7557-3 : 2005, Phần 3: Phương pháp xác định nồng độ cadmi và chì bằng quang phổ hấp thụ nguyên tử ngọn lửa và không ngọn lửa.
LÒ ĐỐT CHẤT THẢI RẮN Y TẾ - XÁC ĐỊNH KIM LOẠI NẶNG TRONG KHÍ THẢI – PHẦN 1: QUY ĐỊNH CHUNG
Health care solid waste incinerators – Determination of heavy metals in fluegas – Part 1: General requirements
Cảnh báo:
- Các nhà hóa học hoặc kỹ thuật viên áp dụng tiêu chuẩn này phải được đào tạo, huấn luyện và có kinh nghiệm.
- Cần chú ý đặc biệt đến độc tính của các kim loại nặng, các dung dịch của chúng và các thuốc thử dùng trong phân tích. Cần cẩn trọng khi sử dụng và thải bỏ các dung dịch sau khi phân tích. Các hóa chất độc bay hơi phải thao tác trong tủ hút độc và không được hút các thuốc thử bằng mồm khi dùng pipet. Cần phòng ngừa các phản ứng giữa chất oxy hóa với chất khử như kali permanganat và H2O2 tạo hỗn hợp nổ.
- Khi làm việc phải sử dụng đầy đủ các phương tiện bảo vệ cá nhân như găng tay, khẩu trang, áo choàng, tạp dề cao su, mặt nạ phòng độc …
1 Phạm vi áp dụng
1.1 Tiêu chuẩn này quy định phương pháp lấy mẫu các kim loại nặng trong bụi và khí thải của lò đốt chất thải rắn nguy hại hoặc các quá trình đốt tương tự, quy về điều kiện nhiệt độ và áp suất tiêu chuẩn.
CHÚ THÍCH: Nhiệt độ và áp suất tiêu chuẩn là nhiệt độ 0 0C và áp suất 101,3 kPa.
Bảng 1 – Giới hạn phát hiện (IDL) các kim loại nặng
|
Kim loại |
ICP-AESa µg/l |
AA ngọn lửab µg/l |
AA cuvet graphitc µg/l |
AA hóa hơia lạnh µg/l |
|
Antimoan/Stibi (Sb) |
40 e |
200 e |
3 e |
|
|
Asen (As) |
60 |
2 f |
1 |
|
|
Bari (Ba) |
2 |
100 |
|
|
|
Beri (Be) |
0,3 |
5 |
0,2 |
|
|
Cadmi (Cd) |
4 |
5 |
0,1 |
|
|
Crom (Cr) tổng |
7 |
50 |
1 |
|
|
Coban (Co) |
7 |
50 |
1 |
|
|
Đồng (Cu) |
6 |
20 |
|
|
|
Chì (Pb) |
50 |
100 |
1 |
|
|
Mangan (Mn) |
2 |
10 |
|
|
|
Thủy ngân (Hg) |
|
|
|
0,2 |
|
Niken (Ni) |
20 |
40 |
|
|
|
Phospho (P) |
60 |
|
|
|
|
Selen (Se) |
80 |
2f |
|
|
|
Bạc (Ag) |
7 |
10 |
|
|
|
Tali (TI) |
40 |
100 |
1 |
|
|
Kẽm (Zn) |
2 |
5 |
|
|
|
CHÚ THÍCH: a Giới hạn phát hiện bằng quang phổ phát xạ nguyên tử (ICP-AES). b Giới hạn phát hiện bằng quang phổ hấp thụ nguyên tử ngọn lửa (F-AAS). c Giới hạn phát hiện bằng quang phổ hấp thụ nguyên tử không ngọn lửa (cuvet graphit) (GF-AAS). d Giới hạn phát hiện bằng quang phổ hấp thụ nguyên tử kỹ thuật hóa hơi lạnh (CV-AAS). e Giới hạn phát hiện với Sb có thể cao hơn phụ thuộc vào cách phá mẫu. f Giới hạn phát hiện As bằng quang phổ hấp thụ nguyên tử kỹ thuật hydrua hóa. |
||||
1.2 Phương pháp này cũng có thể dùng để xác định sự phát thải bụi theo quy trình trình bày ở 7.1.5.2.
1.3 Phương pháp phân tích được nêu trong Bảng 1 và các phép phân tích tương tự thì phép phân tích phổ phát xạ nguyên tử plasma (ICP-AES) có khoảng tuyến tính rộng hơn cả. Những mẫu chứa kim loại có nồng độ từ hàng µg/l đến hàng mg/l đều có thể phân tích được bằng kỹ thuật này. Mẫu chứa crôm, chì hoặc asen cao hơn 50 mg/l cần được pha loãng đến khoảng trên hoặc thấp hơn để phân tích. Mẫu chứa cadmi lớn hơn 20 mg/l cần pha loãng trước khi phân tích.
1.4 Giới hạn phát hiện của phương pháp phụ thuộc vào mẫu và có thể thay đổi theo thành phần mẫu. Giới hạn phát hiện đối với antimoan/stibi cũng phụ thuộc vào cách phá mẫu được dùng và có thể cao hơn số liệu nêu trong Bảng 1. Giới hạn phát hiện cho các kim loại có thể khác biệt với số liệu nêu trong bảng khi phá mẫu bằng HF.
1.5 Sự phức tạp của phương pháp này là nhằm thu được những kết quả tin cậy. Các nhà phân tích cần có kinh nghiệm ở tất cả các bước từ khi lấy mẫu, xử lý, chuẩn bị thuốc thử đến tuân thủ đầy đủ các biện pháp an toàn và sử dụng các phương tiện bảo vệ cá nhân.
2 Tài liệu viện dẫn
Các tài liệu viện dẫn sau là rất cần thiết cho việc áp dụng tiêu chuẩn. Đối với các tài liệu viện dẫn ghi năm ban hành thì áp dụng bản được nêu. Đối với các tài liệu viện dẫn không ghi năm ban hành thì áp dụng phiên bản mới nhất.
TCVN 4851 : 1989 (ISO 3696). Nước dùng để phân tích trong phòng thí nghiệm - Yêu cầu kỹ thuật và phương pháp thử.
TCVN 5977, Sự phát thải của nguồn tĩnh - Xác định nồng độ và lưu lượng bụi trong các ống dẫn khí - Phương pháp khối lượng thủ công.
TCVN 7557 - 2 : 2005, Lò đốt chất thải rắn y tế - Xác định kim loại nặng trong khí thải - Phần 2: Phương pháp xác định nồng độ thủy ngân bằng quang phổ hấp thụ nguyên tử kỹ thuật hóa hơi lạnh.
3 Tóm tắt phương pháp
3.1 Mẫu được lấy đẳng tốc từ nguồn. Đối với mẫu bụi thì lấy bằng đầu lấy mẫu chứa cái lọc được làm nóng, đối với mẫu khí thì lấy mẫu trong dãy bình được làm lạnh được mô tả trong Hình 1 và 4.1.6, hai bình để trống, hai bình chứa dung dịch axit nitric loãng và hydro peroxyt, hai bình khác chứa dung dịch KMnO4 và axit sunfuric, bình cuối cùng chứa chất làm khô.
3.2 Các thành phần trong hệ thống lấy mẫu được thu hồi và phá mẫu riêng theo nửa trước và nửa sau của hệ thống lấy mẫu. Kim loại thu được trong hệ thống lấy mẫu được hòa tan trong axit để các chất vô cơ tan hết còn các chất hữu cơ có thể cản trở việc phân tích bị loại đi.
3.3 Dung dịch axit nitric và hydro peroxyt, dung dịch HCl dùng để tráng, dung dịch KMnO4 trong môi trường axit và dung dịch tráng đầu lấy mẫu và dung dịch phá mẫu trên cái lọc cũng được dùng để phân tích thủy ngân bằng phương pháp phổ hấp thụ nguyên tử hóa hơi lạnh. Toàn bộ bẫy trong hệ thống lấy mẫu, trừ dung dịch kali permanganat, dung dịch tráng HCl và nước để thu thủy ngân, có thể dùng để phân tích các kim loại cho trong Bảng 1 bằng quang phổ hấp thụ nguyên tử ngọn lửa, quang phổ hấp thụ nguyên tử với cuvet graphit.
3.4 Để thuận tiện, phần hút của phần 1A như trình bày ở 7.2.3.2 và phần 2A như trình bày ở 7.2.4 có thể kết hợp lại để phân tích. Phần 1A thường được pha loãng đến 300 ml trước khi phân tích và thêm vào phần 2A được pha loãng đến 150 ml trước khi phân tích. Việc kết hợp cần tuân theo tỷ lệ giữa phần 1A và 2A.
3.5 Hiệu quả của phương pháp phân tích được đánh giá qua mẫu kiểm tra hoặc đo chất lượng như trình bày ở điều 9 của phương pháp này gồm cả kiểm tra hiệu ứng nền.
4 Thiết bị, dụng cụ
Dụng cụ thông thường ở phòng thử nghiệm và các thiết bị sau:
4.1 Hệ thống lấy mẫu
Sơ đồ hệ thống lấy mẫu được mô tả trong Hình 1. Hệ thống lấy mẫu gồm những bộ phận sau:
4.1.1 Đầu lấy mẫu và ống dẫn: làm bằng thạch anh hoặc thủy tinh borosilicat, giống TCVN 7557 - 2 : 2005, trừ đầu lấy mẫu yêu cầu bằng thủy tinh để tránh nhiễm bẩn mẫu gây cản trở cho phân tích.
4.1.2 Ống dẫn và thiết bị đo áp lực
4.1.3 Cái lọc: làm sạch bằng thạch anh hoặc thủy tinh borosilicat không có chất kết dính. Tạp chất trong cái lọc phải ít hơn 0,186 µg/cm2 cho mỗi kim loại cần đo. Cái lọc cần có hiệu quả lọc 99,95 % (< 0,05="" %="" thấm="" qua)="" với="" khói="" dioctyl="" phtalat="" 0,3="" µg.="" để="" xác="" định="" bụi="" trong="" nguồn="" chứa="">2 hoặc SO3, chất liệu làm cái lọc phải không được phản ứng với SO2 hoặc SO3 như TCVN 7557 - 2 : 2005. Nên dùng sợi thạch anh.
4.1.4 Giá đỡ cái lọc: bằng thủy tinh như TCVN 7557-2 : 2005, trừ trường hợp giá đỡ bằng teflon hoặc không bằng kim loại, không bị nhiễm bẩn.
4.1.5 Hệ thống sấy nóng cái lọc: theo TCVN 7557-2 : 2005.
4.1.6 Bình ngưng
4.1.6.1 Hệ thống ngưng tụ sau đây phải được dùng để ngưng tụ và thu hơi kim loại và xác định độ ẩm trong khí ống khói. Hệ thống ngưng tụ gồm ba đến bảy bình nối liên tiếp với nhau bằng thủy tinh nhám hoặc thứ khác không dò, không gây nhiễm bẩn. Bình teflon phải có cùng hình dạng và kích thước với bình lấy mẫu (impinger) bằng thủy tinh có cái nối không dò rỉ, không gây nhiễm bẩn mẫu. Ngoài ra khoảng cách từ đáy ống dẫn khí của bình teflon tới chỗ nối với bình chứa dung dịch axit phải bằng với khoảng cách đó của bình thủy tinh. Bình đầu tiên là tùy chọn và bình này đóng vai trò một bẫy ẩm trong quá trình thử nghiệm.
Bình đầu tiên để trống. Bình thứ hai và thứ ba chứa lượng đã biết dung dịch axit nitric và hydro peroxyt. Bình thứ tư trống. Bình thứ năm và sáu chứa lượng đã biết dung dịch kali permanganat và bình cuối cùng chứa một lượng đã biết silicagen hoặc chất làm khô khác. Ở lối ra của bình cuối cùng đặt một nhiệt kế đo được đến 1 0C (2 0F).
4.1.6.2 Bình đầu tiên dùng làm bẫy ẩm như TCVN 7557-2 : 2005. Bình thứ hai (hoặc bình thứ nhất chứa HNO3/H2O2) giống bình thứ nhất như TCVN 7557-2 : 2005. Bình thứ ba (bình HNO3/H2O2 thứ hai) giống bình Greenburg - Smith thứ hai như TCVN 7557-2 : 2005. Tất cả các bình trong hệ thống lấy mẫu kim loại đều giống bình thứ nhất HNO3/H2O2.
4.1.6.3 Tùy theo điều kiện lấy mẫu, bình đầu tiên có thể bỏ qua nếu hơi ẩm ngưng tụ ít hơn khoảng 100 ml. Khi không cần bình bẫy ẩm thì bỏ nó ra khỏi hệ thống, còn các bình khác giữ nguyên. Nếu không phân tích thủy ngân thì bình chứa KMnO4 và bình để trống sau đó có thể bỏ đi.
4.1.7 Hệ thống đo, áp kế và thiết bị xác định mật độ của khí: theo TCVN 7557-2 : 2005.
4.1.8 Băng teflon: dùng trong các mối nối của hệ thống.
4.1.9 Bơm lấy mẫu có tốc độ hút từ 1 l/min đến 5 l/min với ống nối mềm.
4.1.10 Thùng vận chuyển: được làm lạnh bằng đá khô (đá CO2).
4.2 Thiết bị thu mẫu
Giống như TCVN 7557-2 : 2005 nhưng có những ngoại lệ sau:
4.2.1 Ống dẫn mẫu không kim loại, bàn chải và khăn lau đầu lấy mẫu: nhằm định lượng các chất ở nửa trước của hệ thống. Vật liệu của bàn chải và khăn lau bằng teflon.
4.2.2 Bình chứa mẫu: bình thủy tinh 1000 ml đến 500 ml, có nắp teflon không phản ứng với các dung dịch oxy hóa, được dùng cho mẫu và mẫu trắng chứa KMnO4. Bình polyetylen có thể dùng cho các loại mẫu khác.
4.2.3 Kẹp polyetylen và găng tay plastic: dùng để lấy cái lọc từ giá đỡ trong hệ thống.
4.3 Chuẩn bị mẫu và thiết bị phân tích
Đọc phần chuẩn bị mẫu và kỹ thuật phân tích. Xem 7.2 về kỹ thuật chuẩn bị mẫu.
5 Thuốc thử
Trong phân tích, trừ khi có những quy định khác, chỉ sử dụng thuốc thử có độ tinh khiết được thừa nhận.
CHÚ THÍCH: Vì nồng độ axit ảnh hưởng tới việc đo độ hấp thụ của các kim loại nên các dung dịch đó, kể cả dung dịch tiêu chuẩn và dung dịch trắng đều phải có cùng nồng độ axit.
5.1 Nước cất hoặc nước đã loại khoáng hoặc nước có độ tinh khiết tương đương
Theo TCVN 4851 : 1989 (ISO 3696).
5.2 Axit clohydric đặc (HCl) p = 1,16 g/ml (37%)
5.3 Axit nitric đặc (HNO3) p = 1,42 g/ml
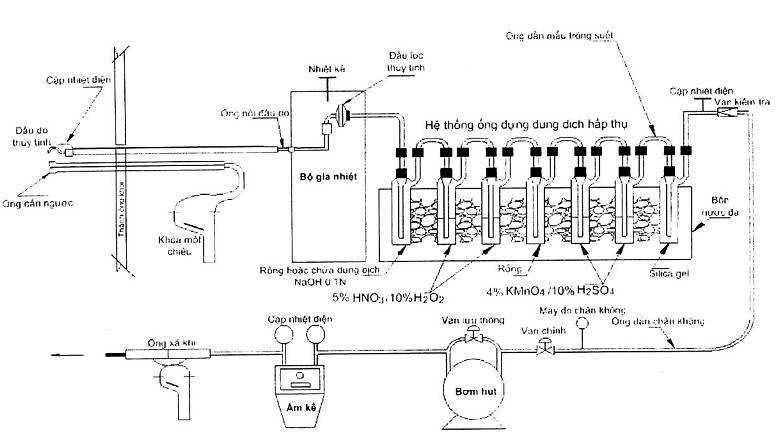
Hình 1 - Sơ đồ hệ thống lấy mẫu các kim loại nặng
5.4 Axit nitric (0,1 M): Vừa thêm vừa khuấy 6,3 ml axit nitric đặc vào bình chứa khoảng 900 ml nước. Pha loãng đến 1000 ml bằng nước. Trộn đều. Dung dịch phải ít hơn 2 µg/l đối với mỗi kim loại.
5.5 Axit nitric 10 % (thể tích): Vừa thêm vừa khuấy 500 ml HNO3 đặc vào bình chứa khoảng 4000 ml nước. Pha loãng đến 5000 ml bằng nước. Trộn đều. Dung dịch phải ít hơn 2 µg/l đối với mỗi kim loại.
5.6 Axit nitric 5 % (thể tích): Vừa thêm vừa khuấy 50 ml HNO3 đặc vào 800 ml nước. Pha loãng đến 1000 ml bằng nước. Dung dịch phải ít hơn 2 µg/l đối với mỗi kim loại.
5.7 Axit nitric 50 % (thể tích): Vừa thêm vừa khuấy 125 ml HNO3 đặc vào bình chứa khoảng 100 ml nước. Pha loãng đến 250 ml bằng nước. Trộn đều. Dung dịch phải ít hơn 2 µg/l đối với mỗi kim loại.
5.8 Dung dịch hấp thụ HNO3 5 % (thể tích)/ H2O2 10 % (thể tích): Vừa thêm cẩn thận vừa khuấy 50 ml HNO3 đặc vào bình định mức 1000 ml có chứa 500 ml nước. Thêm cẩn thận 333 ml H2O2 30 % vào bình. Thêm nước đến vạch. Dung dịch phải ít hơn 2 µg/l đối với mỗi kim loại.
5.9 Axit HCl 8 M: Thêm cẩn thận, vừa thêm vừa khuấy 690 ml HCl đặc vào bình chứa 250 ml nước. Pha loãng đến 1000 ml bằng nước. Trộn đều. Dung dịch phải ít hơn 2 µg/l Hg.
5.10 Hydro peroxyt đặc 30 % (thể tích).
5.11 Kali permanganat 5 % (khối lượng trên thể tích).
5.12 Dung dịch hấp thu KMnO4 4 % (khối lượng trên thể tích) + H2SO4 10 % (thể tích)
Trộn cẩn thận 100 ml H2SO4 đặc vào 800 ml nước. Vừa thêm nước đến 1000 ml vừa khuấy. Dung dịch này là H2SO4 10 % (thể tích). Hòa tan khi khuấy 40 g KMnO4 vào dung dịch H2SO4 10 %. Thêm nước đến 1 lít. Pha và giữ trong bình thủy tinh để tránh phân hủy. Dung dịch phải ít hơn 2 µg Hg/l.
Chuẩn bị dung dịch hàng ngày.
CHÚ THÍCH: Để giảm sự tự phân hủy của dung dịch permanganat, lọc dung dịch qua giấy lọc What - man 541. Do phản ứng của KMnO4 với axit sinh khí trong bình chứa nên các bình này không được đổ đầy và cần thông hơi để tránh nổ. Thông hơi là cần nhưng tránh để dung dịch bị nhiễm bẩn. Nên dùng nắp có khoan lỗ 70 - 72 và ống dẫn bằng teflon.
5.13 Axit sunfuric (H2SO4) đặc p = 1,84 g/ml (98%)
5.14 Silicagen và nước đá: theo TCVN 7557-2 : 2005.
5.15 Axit flohydric HF đặc
6 Lấy mẫu, lưu giữ và xử lý mẫu
6.1 Lấy mẫu
6.1.1 Chuẩn bị trước khi thử nghiệm: Theo phương pháp chung cho ở TCVN 7557-2 : 2005 nhưng cái lọc không cần làm khô và cân, trừ trường hợp cần phân tích bạc. Dụng cụ thủy tinh của hệ thống đầu tiên được tráng bằng vòi nước nóng, sau đó rửa bằng nước xà phòng nóng. Rồi tráng ba lần bằng nước rồi ba lần bằng nước cất. Dụng cụ thủy tinh đem ngâm vào dung dịch HNO3 10 % (thể tích) ít nhất 4 h, tráng ba lần bằng nước cất, lần cuối cùng bằng axeton rồi để khô trong không khí. Cần đậy kín để tránh nhiễm bẩn.
6.1.2 Chuẩn hóa hệ thống: Chuẩn hóa hệ thống theo TCVN 7557-2 : 2005 : đầu lấy mẫu; ống dẫn; hệ thống đo; bộ phận đốt nóng đầu lấy mẫu; nhiệt kế; kiểm tra độ kín của hệ thống đo; áp kế.
6.1.3 Xác định sơ bộ: Theo TCVN 7557-2 : 2005.
6.1.4 Chuẩn bị hệ thống lấy mẫu
6.1.4.1 Theo phương pháp chung cho ở TCVN 7557-2 : 2005. Thêm 100 ml HNO3/H2O2 (điều 5.8) vào mỗi bình chứa HNO3/ H2O2 (là bình thứ hai và thứ ba) như mô tả trên Hình 1. Thêm 100 ml dung dịch hấp thụ kali permanganat trong môi trường axit (điều 5.12) vào mỗi bình chứa kali pecmanganat trong môi trường axit (hai bình). Lấy 200 g đến 300 g silicagen cho vào bình cuối. Có thể cân silicagen trực tiếp trong bình chứa trước khi lắp thiết bị.
6.1.4.2 Có nhiều cách lựa chọn dựa trên điều kiện lấy mẫu. Không cần bình rỗng đầu tiên nếu lượng ẩm trong bình theo tính toán hoặc được xác định là nhỏ hơn 100 ml.
6.1.4.3 Giữ dung dịch HNO3/H2O2 và 100 ml kali pecmanganat trong môi trường axit làm dung dịch trắng. Những dung dịch trắng này cần ghi nhãn và phân tích theo điều 7. Lắp hệ thống lấy mẫu như Hình 1, chú ý không để dò rỉ ở những chỗ nối, nên dùng băng teflon thay cho mỡ silicon để tránh gây nhiễm bẩn.
CHÚ THÍCH: Cần hết sức cẩn thận để tránh nhiễm bẩn trong hệ thống lấy mẫu. Thuốc thử để hấp thụ Hg (kali pecmanganat trong môi trường axit) có thể gây nhiễm bẩn khi phân tích mangan. Cần tránh trộn lẫn giữa H2O2 và kali pecmanganat trong môi trường axit.
6.1.4.4 Có thể phân tích riêng Hg bằng một hệ thống lấy mẫu riêng theo TCVN 7557-2.
6.1.5 Kiểm tra độ kín: Theo TCVN 7557-2 : 2005: kiểm tra trước, kiểm tra trong khi lấy mẫu, kiểm tra sau khi lấy mẫu.
6.1.6 Vận hành hệ thống lấy mẫu: Theo TCVN 7557-2 : 2005. Ghi chép theo TCVN 7557-2 : 2005. Khi lấy mẫu thủy ngân, sử dụng phương pháp tương tự như Phụ lục B TCVN 7557-2 : 2005, nếu cần, duy trì màu hồng của pemanganat ở bình cuối.
6.1.7 Tính phần trăm tốc độ lấy mẫu đẳng tốc: Theo TCVN 7557-2 : 2005.
7 Cách tiến hành
7.1 Thu mẫu
Khi lấy mẫu xong, rút ngay đầu lấy mẫu ra khỏi ống khói và thu mẫu.
7.1.1 Cần để nguội đầu lấy mẫu trước khi thu hồi. Quét sạch bụi ở ngoài đầu lấy mẫu và đậy mũi lấy mẫu bằng nắp sạch để tránh mất hoặc lẫn thêm bụi vào mẫu. Không đậy mũi lấy mẫu quá kín khi để nguội, điều đó có thể gây ra sự hút dung dịch từ bình lên cái lọc.
7.1.2 Trước khi chuyển hệ thống lấy mẫu tới nơi làm sạch, tháo dây khỏi bình cuối và đậy bình bằng nắp sạch làm bằng thủy tinh nhám, nắp plastic hoặc bằng teflon. Đậy nắp đường ra và đường vào của bình.
7.1.3 Có thể tháo hệ thống lấy mẫu trước khi giá lọc/lò hoàn toàn nguội.
Cách làm như sau: trước tiên tháo giá đỡ cái lọc ra/bình impinger nới lỏng nắp ở đầu phía cuối. Sau đó tháo đầu lấy mẫu khỏi giá lọc vào và nới lỏng nắp đầu ở phía cuối. Đậy nắp đầu lấy mẫu và dây dẫn như đã trình bày ở trên.
7.1.4 Chuyển đầu lấy mẫu chứa cái lọc và bình hấp thụ đến nơi làm sạch, ở đó sẽ tiến hành làm sạch và cần tránh gió để tránh mất mẫu cũng như nhiễm bẩn mẫu. Kiểm tra hệ thống lấy mẫu trước và trong khi tháo, ghi lại những điều bất thường.
7.1.5 Thu mẫu và xử lý theo sơ đồ trên Hình 2.
Không để những dụng cụ dùng để thu mẫu gây nhiễm bẩn mẫu.
7.1.5.1 Bình chứa N0 1 (cái lọc). Lấy cẩn thận cái lọc khỏi giá đỡ và đặt vào đĩa petri. Dùng kẹp bọc polypropylen hoặc teflon đã được rửa bằng axit hoặc găng tay sạch để làm việc với cái lọc. Nếu cần gấp cái lọc phải đảm bảo bụi nằm trong cái lọc. Chuyển cẩn thận cái lọc và bụi hoặc sợi lọc vào đĩa petri bằng bàn chải nylông khô, cứng. Không được dùng vật liệu chứa kim loại khi thu mẫu. Dán nhãn cho hộp petri.
CHÚ THÍCH: Theo điều 7.1.5.2 chỉ khi xác định sự phát thải bụi kèm phát thải kim loại. Nếu chỉ cần xác định phát thải kim loại thì bỏ qua 7.1.5.2 và tiến hành theo 7.1.5.3.
7.1.5.2 Bình chứa N0 2 (tráng bằng axeton)
7.1.5.2.1 Cẩn thận tránh để bụi ở ngoài đầu lấy mẫu rơi vào mẫu. Thu để định lượng chất rắn và chất lỏng từ mũi lấy mẫu, đầu lấy mẫu, vòng đệm (vòng đệm làm bằng plastic như teflon, polypropylen… để phòng ngừa nhiễm bẩn từ vòng đệm kim loại, có thể dùng vòng đệm thủy tinh nhưng phương pháp này không yêu cầu), ống dẫn, nửa trước của giá lọc bằng cách rửa bằng 100 ml axeton và thu vào bình thủy tinh. Cần dùng chính xác 100 ml vì còn hiệu chỉnh mẫu trắng. Có thể dùng nước cất thay cho axeton nếu được phép, trường hợp này cần tiết kiệm phần nước làm mẫu trắng. Cách rửa bằng axeton như sau: tháo cẩn thận mũi lấy mẫu và tráng mặt trong bằng axeton bằng bình rửa và chải bằng bàn chải không kim loại. Chải đến khi axeton không vẩn bụi, sau đó tráng mặt trong lần nữa bằng axeton. Tương tự, chải sạch và tráng vòng đệm Swagelok bằng axeton.
7.1.5.2.2 Tráng ống dẫn của đầu lấy mẫu bằng axeton bằng cách nghiêng và quay đầu lấy mẫu trong khi phun axeton vào phần trên của nó sao cho toàn bộ mặt trong tiếp xúc với axeton. Thu axeton vào bình chứa. Có thể dùng phễu để chuyển nước rửa vào bình chứa. Tiếp theo là cọ bằng bàn chải. Giữ đầu lấy mẫu ở vị trí nghiêng, tia axeton từ phía bên trong khi chải xoắn theo đầu lấy mẫu; giữ bình chứa bên dưới đầu lấy mẫu để thu hết axeton và chất rắn. Tráng và chải ba lần hoặc nhiều hơn để không còn thấy cặn trong axeton và ống dẫn của đầu lấy mẫu. Tráng bàn chải bằng axeton và gộp vào bình chứa.
7.1.5.2.3 Nên có hai người rửa đầu lấy mẫu để tránh mất mẫu. Giữa các lần lấy mẫu cần giữ cho bàn chải sạch và không bị nhiễm bẩn.
7.1.5.2.4 Rửa sạch nửa trước của giá đỡ cái lọc bằng axeton và bàn chải cứng. Tráng mỗi bề mặt ba lần hoặc nhiều hơn khi không còn nhìn thấy các hạt bằng mắt thường. Tráng bàn chải và giá đỡ cái lọc lần cuối. Sau khi thu axeton và chất rắn vào bình mẫu, nút kín bình để axeton không thoát ra ngoài khi vận chuyển đến phòng thí nghiệm. Đánh dấu mức chất lỏng để xem có bị dò rỉ trong khi vận chuyển hay không. Dán nhãn.
7.1.5.3 Bình chứa N0 3 (tráng đầu lấy mẫu). Giữ đầu lấy mẫu theo 7.1.5.2 khi tráng bằng HNO3 0,1 M như sau: tráng cẩn thận ống dẫn của đầu lấy mẫu, mũi lấy mẫu, cái lọc và phần trước của giá đỡ cái lọc bằng 100 ml HNO3 0,1 M và thu vào bình chứa mẫu.
CHÚ THÍCH: cần thiết dùng chính xác 100 ml để hiệu chỉnh tráng tiếp theo. Ghi tổng thể tích dung dịch tráng. Đánh dấu mức chất lỏng ở ngoài bình chứa để xem có bị dò rỉ trong quá trình vận chuyển. Đậy kín bình và dán nhãn rõ ràng. Cuối cùng tráng mũi lấy mẫu, ống dẫn của đầu lấy mẫu, nửa trước của giá lọc bằng nước, axeton và loại bỏ các dịch tráng này.
7.1.5.4 Bình chứa N04 (Bình 1 đến 3, bình HNO3/H2O2 và bình đo độ ẩm nếu có, và dung dịch tráng). Vì lượng chất lỏng lớn nên dung dịch ở các bình 1, 2 và 3 có thể được chứa trong nhiều bình. Đo chất lỏng trong ba bình đầu tiên chính xác đến 0,5 ml bằng ống đong hoặc cân chính xác đến 0,5 g. Ghi thể tích đó lại. Thông tin này cần để tính độ ẩm của khí ống khói. Rửa ba bình, giá đỡ cái lọc, nửa sau của giá đỡ cái lọc, các khớp nối thủy tinh cẩn thận bằng 100 ml HNO3.
CHÚ THÍCH: Dùng chính xác 100 ml HNO3 là cần thiết để hiệu chỉnh trắng. Dung dịch trong bình và dung dịch rửa được gộp lại, đo và ghi thể tích. Đánh dấu mức chất lỏng để xác định sự dò rỉ khi vận chuyển. Nút kín bình chứa và dán nhãn.
7.1.5.5 Bình chứa N0 5A (HNO3 0,1 m), 5B (KMnO4/H2SO4) và 5C (dung dịch tráng và pha loãng HCl 8 M). Nếu không xác định Hg thì các bình 4, 5, 6 trong Hình 1 có thể bỏ đi.
7.1.5.5.1 Đổ toàn bộ chất lỏng trong bình trống (khi bắt đầu lấy mẫu) và tiếp theo là hai bình permanganat) vào ống đong và đo thể tích chính xác đến 0,5 ml. Thông tin này cần để tính độ ẩm của khí ống khói. Chuyển chất lỏng vào bình chứa 5A. Tráng bình số 4 bằng 100 ml HNO3 0,1 M và chuyển vào bình chứa N0 5A. Rót chất lỏng của hai bình chứa kali pecmanganat trong môi trường axit vào bình chứa N0 5B. Dùng 100 ml dung dịch kali pecmanganat trong môi trường axit mới tráng các bình kali pecmanganat trong môi trường axit và các chỗ nối thủy tinh ba lần. Thu vào bình chứa N0 5B, chú ý không để mất kết tủa trong các bình permanganat. Dùng 100 ml nước tráng các bình kali pecmanganat trong môi trường axit và các khớp nối thủy tinh ba lần và thu vào bình chứa N0 5B, chú ý không để mất kết tủa trong các bình permanganat. Đánh dấu mức chất lỏng để xác định sự dò rỉ khi vận chuyển. Dán nhãn.
CHÚ THÍCH - Do phản ứng của KMnO4 với axit nên có thể sinh khí trong bình chứa. Bởi vậy các bình chứa này không đổ đầy và cần thông hơi. Nên khoan lỗ 70 - 72 ở nắp bình và dùng ống dẫn teflon.
7.1.5.5.2 Nếu không còn kết tủa sau khi tráng bằng nước thì không cần tráng bằng HCl. Tráng bằng nước để làm sạch permanganat, nhưng nếu có thể kết tủa thì cần làm sạch ở nơi thoáng hoặc trong tủ hút để tránh khí clo. Nếu kết tủa còn lại sau khi tráng bằng nước thì rửa thành bình bằng 25 ml HCl 8 M và thu vào bình chứa N0 5C đã có sẵn 200 ml nước. Rửa thành bình bằng cách quay để HCl tiếp xúc với toàn thể mặt trong. Dùng 25 ml HCl 8 M để tráng cả hai bình permanganat. Tráng bình thứ nhất, sau đó đổ dung dịch sang bình thứ hai. Cuối cùng gộp toàn bộ 25 ml HCl 8 M vào bình chứa 5C. Đánh dấu mức chất lỏng để xác định dò rỉ khi vận chuyển.
7.1.5.6 Bình chứa N0 6 (silicagen). Ghi màu của chỉ thị silicagen để xác định nó đã được dùng hết chưa, chuyển silicagen vào bình ban đầu và gắn kín lại. Có thể dùng phễu và găng tay để chuyển silicagen. Không cần loại đi một ít silicagen vụn có thể bám lại ở thành bình. Không dùng nước hoặc chất lỏng khác để chuyển silicagen vì khối lượng của silicagen được dùng cho tính độ ẩm. Có thể cân ngay silicagen cùng với bình bằng cân có độ chính xác đến 0,5 g ở hiện trường.
7.1.5.7 Bình chứa N0 7 (axeton trắng). Nếu cần xác định bụi, điều đó cần làm ít nhất 1 lần, lấy 100 ml axeton dùng trong quá trình thu mẫu vào bình chứa có nhãn sẵn để làm mẫu trắng. Đậy kín bình.
7.1.5.8 Bình chứa N0 8A (HNO3 0,1 trắng). Làm ít nhất một lần trong khi thử, lấy 300 ml HNO3 0,1 M dùng trong quá trình thu mẫu vào bình có nhãn sẵn để làm dung dịch trắng. Đậy kín bình.
7.1.5.9 Bình chứa N0 8B (nước trắng). Làm ít nhất một lần trong khi thử, lấy 100 ml nước dùng trong quá trình thu mẫu vào bình N0 8B. Đóng kín bình.
7.1.5.10 Bình chứa N0 9 (HNO3 5 %/H2O2 10 % trắng). Làm ít nhất một lần trong khi thử, lấy 200 ml dung dịch HNO3 5 % và H2O2 10 % vào bình N0 9. Đậy kín bình.
7.1.5.11 Bình chứa N0 10 (KMnO4/H2SO4 trắng). Làm ít nhất một lần trong khi thử, lấy 100 ml dung dịch KMnO4/H2SO4 dùng cho quá trình thu mẫu vào bình N0 10 để làm mẫu trắng cho phân tích Hg. Chuẩn bị bình chứa như chú thích ở 7.2.5.5.1.
7.1.5.12 Bình chứa N0 11 (HCl 8 M trắng). Làm ít nhất một lần trong khi thử, lấy 200 ml nước cho vào bình N0 11 rồi rót vào đó 25 ml HCl 8 M trong khi khuấy.Trộn đều và đóng kín bình.
7.1.5.13 Bình chứa N0 12 (cái lọc trắng). Làm ít nhất một lần trong quá trình thử, lấy một cái lọc cùng loại với cái lọc đã dùng đặt vào đĩa petri. Đậy đĩa petri. Đó là cái lọc trắng.
7.2 Chuẩn bị mẫu
Ghi mức chất lỏng ở mỗi bình chứa vào sổ phân tích, xác định sự dò rỉ khi vận chuyển. Nếu có dò rỉ thì có thể bỏ mẫu đó hoặc phải hiệu chỉnh kết quả. Hình 3 mô tả biểu đồ về sự chuẩn bị mẫu và phương pháp phân tích cho mỗi thành phần mẫu. Nếu hệ thống lấy mẫu dùng là dạng cyclon thì bộ phận thu bụi phải được chuẩn bị và xử lý bằng phương pháp tương tự như với cái lọc và được gộp chung vào mẫu đã phá của cái lọc. Phá mẫu bằng axit.
7.2.1 Bình chứa N0 1 (cái lọc). Nếu cần xác định phát thải bụi thì làm khô cái lọc và cái bẫy lọc đến khối lượng không đổi. Để phân tích kim loại thì chia cái lọc và bẫy lọc thành nhiều phần, mỗi phần khoảng 0,5 g đặt vào bình chịu áp lực hoặc bình phá mẫu chịu áp lực. Thêm vào mỗi bình 6 ml HNO3 đặc và 4 ml HF đặc. Phá mẫu bằng vi sóng, hoặc đặt vào bình phá mẫu chịu áp lực ở 140 0C (285 0F) trong 6 h. Để nguội mẫu đến nhiệt độ phòng và trộn với phần tráng đầu lấy mẫu đã phá theo 7.2.3.
Cảnh báo: HF rất độc. Cần cẩn thận khi làm việc với HF.
7.2.2 Bình chứa N0 2 (axeton trắng). Đo chất lỏng trong bình chứa này theo thể tích chính xác đến ± 1 ml hoặc khối lượng đến ± 0,5 g. Chuyển sang cốc 250 ml đã tráng axit và cân trước rồi cho bay hơi đến khô ở nhiệt độ và áp suất thường. Nếu cần phân tích bụi thì làm khô bằng chất làm khô trong 24 h (không dùng nhiệt) đến khối lượng không đổi theo phương pháp xác định thủy ngân và công bố kết quả chính xác đến 0,1 mg. Hòa tan cặn bằng 10 ml HNO3 đặc và nhập toàn bộ chất lỏng và rắn vào bình chứa N0 3 vừa làm vừa khuấy và bắt đầu làm theo điều 7.2.3
7.2.3 Bình chứa N0 3 (tráng đầu lấy mẫu). pH của mẫu phải là 2 hoặc thấp hơn. Nếu pH lớn hơn 2 phải thêm HNO3 một cách cẩn thận, khi khuấy, đến pH 2. Mẫu được chuyển vào cốc chứa nước và được đậy bằng nắp kính đồng hồ. Thể tích mẫu được cô trên bếp ở nhiệt độ thấp hơn nhiệt độ sôi đến khi còn 20 ml và phá mẫu bằng một trong cách sau:
7.2.3.1 Phá mẫu theo phương pháp thích hợp (vi sóng, bình phá mẫu chịu áp lực ở 140 0C), dùng HF và sau đó làm theo điều 7.2.1.
7.2.3.2 Trộn mẫu này với mẫu đã phá ở 7.2.1. Mẫu kết hợp là Phần 1 - Lọc dung dịch kết hợp giữa dung dịch phá mẫu cái lọc và dung dịch tráng đầu lấy mẫu qua giấy lọc Whatman 541. Pha loãng đến 300 ml (hoặc thể tích thích hợp tùy theo nồng độ kim loại) bằng nước. Dung dịch sau khi pha loãng là Phần 1. Đo và ghi thể tích của Phần 1 chính xác đến 0,1 ml. Lấy chính xác 50 ml và gọi là Phần 1B. Đánh dấu 250 ml còn lại là Phần 1A. Phần 1A được dùng cho phân tích phổ phát xạ nguyên tử plasma, khối phổ plasma hoặc quang phổ hấp thụ nguyên tử cho tất cả các kim loại, trừ Hg. Phần 1B dùng để xác định Hg ở nửa trước của hệ thống lấy mẫu.
7.2.4 Bình chứa N0 4 (bình 1-3). Đo và ghi thể tích tổng của mẫu (Phần 2) chính xác đến 0,5 ml. Lấy 75 ml đến 100 ml để phân tích Hg, gọi là Phần 2B. Đánh dấu phần còn lại là Phần 2A. Toàn bộ Phần 2A được phá mẫu và cô cạn, phần cô này dùng để phân tích các kim loại trừ Hg. Phần 2B được chuẩn bị và phân tích theo điều 7.4.7. Phần 2A có pH 2 hoặc nhỏ hơn, nếu cần, điều chỉnh bằng HNO3 đến pH = 2. Chuyển vào cốc chứa nước và đậy bằng nắp kính đồng hồ. Cô trên bếp ở nhiệt độ thấp hơn nhiệt độ sôi đến thể tích 20 ml. Phá mẫu bằng axit theo phương pháp mô tả trong điều này hoặc theo một trong các phương pháp được mô tả trong Phụ lục E, F, G của TCVN 7557-2 : 2005.
7.2.5 Bình chứa N0 5A, 5B và 5C (bình 4, 5 và 6). Để riêng các mẫu này.
7.2.5.1 Đo và ghi thể tích 5A và 5B chính xác đến 0,5 ml. Pha loãng 5C đến 500 ml bằng nước. Như vậy được Phần 3A, 3B và 3C. Phân tích các phần này theo 7.4.
7.2.5.2 Vì dung dịch tráng kali pecmanganat trong môi trường axit và nước chứa lượng lớn Hg từ bình permanganat, lượng Hg trong dung dịch tráng HCl (Phần 3C) có thể rất nhỏ. Tuy nhiên, như đã nói ở phần đầu của phương pháp này, cộng phần Hg ở Phần 3C vào Phần 1B, 2B, 3A và 3B để tính Hg tổng số.
7.2.6 Bình chứa N0 6 (Silicagen). Cân silicagen đã dùng (hoặc silicagen cùng với bình) chính xác đến 0,5 g bằng cân. (Giai đoạn này có thể làm tại hiện trường).
7.3 Chuẩn hóa
7.4 Phân tích mẫu
7.4.1 Với mỗi hệ thống lấy mẫu cần phân tích bảy bình riêng. Một sơ đồ nhận mẫu, chuẩn bị và phân tích được mô tả ở Hình 3. Hai mẫu đầu, gọi là Phần 1A và 1B, gồm các mẫu hòa tan nửa trước hệ thống. Phần 1A dùng để phân tích quang phổ phát xạ nguyên tử plasma và quang phổ hấp thụ nguyên tử như 7.4.5. Phần 1B dùng để phân tích Hg ở nửa trước như 7.4.5. Phần 1B dùng để phân tích Hg ở nửa trước như 7.4.7.
7.4.2 Nửa sau của hệ thống dùng bình thứ bảy để chuẩn bị mẫu thứ ba. Mẫu thứ ba và bốn, dán nhãn 2A và 2B, chứa mẫu nước do độ ẩm và HNO3/H2O2 từ bình 1 đến bình 3. Phần 2A dùng cho phân tích quang phổ phát xạ nguyên tử plasma và quang phổ hấp thụ nguyên tử. Phần 2B dùng để phân tích Hg.
7.4.3 Mẫu 5A, 5B và 5C được dán nhãn Phần 3A, 3B và 3C. Chúng chứa chất và dung dịch tráng của bình trống 4 và kali pecmanganat trong môi trường axit ở bình 5 và 6. Các mẫu này dùng để phân tích Hg như 7.4.7. Thủy ngân tổng số ở nửa sau của hệ thống là tổng số từ các phần 2B, Phần 3A, 3B và 3C.
7.4.4 Đầu tiên phân tích tất cả các mẫu cho các kim loại trừ Hg. Nếu có sắt và nhôm thì các mẫu cần được pha loãng để nồng độ Fe, Al nhỏ hơn 50 ppm để giảm cản trở đối với asen, cadmi, crom và chì.
CHÚ THÍCH: Khi phân tích mẫu trong HF cần dùng đến Al. Các mẫu ở nửa trước hệ thống đều chứa HF cần dùng đến Al.
7.4.5 ICP - AES (quang phổ phát xạ nguyên tử plasma). Phần 1A và 2A được phân tích bằng ICP - AES. Dùng phương pháp 6010.
7.4.6 AA (quang phổ hấp thụ nguyên tử) hút trực tiếp hoặc dùng cuvet graphit. Nếu phân tích Phần 1A và 2A dùng cuvet graphit hoặc hút trực tiếp thì cần xem bảng A-2 để biết những cản trở có thể có và kỹ thuật để giảm thiểu.
7.4.7 AA phân tích Hg bằng nguyên tử hóa hơi lạnh. Phần 1B, 2B, 3A, 3B và 3C dùng để phân tích riêng rẽ Hg bằng quang phổ hấp thụ nguyên tử kỹ thuật hóa hơi lạnh.
Nếu không biết trước khoảng nồng độ Hg thì pha loãng 1 ml đến 10 ml mẫu đầu thành 100 ml. Ghi rõ số mililit đã dùng để pha loãng thành 100 ml.
Để xác định Hg trong phát thải, lượng mẫu cần lấy để pha loãng phụ thuộc nồng độ Hg: lượng mẫu cần lấy để pha loãng và phân tích phải dưới 1 µg Hg và nằm trong đường chuẩn (từ 0 ng đến 1000 ng).
7.5 Tính kết quả
7.5.1 Thể tích khí khô. Dùng số liệu từ phép thử này để tính Vm(std), thể tích mẫu khí khô ở điều kiện tiêu chuẩn như cho ở 6.3 phương pháp xác định thủy ngân.
7.5.2 Thể tích hơi nước và độ ẩm. Dùng các dữ liệu thu được ở phép thử này để tính thể tích hơi nước Vw(std) và độ ẩm Bws của ống khói. Dùng phương trình 5-2 và 5-3 phương pháp xác định thủy ngân.
7.5.3 Tốc độ khí trong ống khói. Dùng số liệu của phép thử này và phương trình 2-9 của phương pháp 2 tính tốc độ khí trung bình trong ống khói.
7.5.4 Kim loại (trừ Hg) trong mẫu
7.5.4.1 Phần 1A, nửa trước, kim loại (trừ Hg). Tính toán lượng từng kim loại trong Phần 1 của hệ thống lấy mẫu theo phương trình:
Mfh = Ca1 x Fd x Vsoln 1 (1)1)
trong đó:
Mfh là khối lượng tổng của mỗi kim loại (trừ Hg) thu được ở nửa trước của hệ thống (Phần 1), tính bằng microgram;
CM là nồng độ của kim loại trong Phần 1A, tra từ đường chuẩn, tính bằng microgram trên mililit;
Fd là hệ số pha loãng (Fd = nghịch đảo của phân số dung dịch đặc và dung dịch thực dùng khi đọc được nồng độ Ca1, thí dụ, khi pha loãng 2 ml dung dịch phần 1A thành 10 ml thì Fd = 5).
Vsol.1 là thể tích tổng của dung dịch mẫu được phá (Phần 1), tính bằng mililit.
7.5.4.2 Phần 2A, nửa sau, kim loại (trừ Hg). Tính toán lượng mỗi kim loại trong phần 2 theo phương trình:
Mbh = Ca2 x Fa x Va (2)1)
trong đó:
Mbh là khối lượng tổng từng kim loại (trừ Hg) thu được ở nửa sau của hệ thống lấy mẫu (Phần 2), tính bằng microgam;
Ca2 là nồng độ của kim loại ở mẫu chưa pha loãng Phần 2A, tra từ đường chuẩn tính bằng microgram trên mililit;
Fa là hệ số phần hút, thể tích Phần 2 chia cho thể tích phần hút Phần 2A. Xem 7.2.4;
Va là thể tích tổng số của dung dịch mẫu được phá (Phần 2A đặc), tính bằng mililit. Xem 7.2.4.
7.5.4.3 Tổng số kim loại (trừ Hg). Tính tổng lượng kim loại trong hệ thống lấy mẫu như sau:
Mt = (Mfh - Mfhb) + (Mbh - Mbnb) (3)1)
trong đó:
Mt là khối lượng tổng các kim loại thu được trong hệ thống lấy mẫu, tính bằng microgam;
Mfhb là giá trị hiệu chỉnh trắng của kim loại ở nửa trước, tính bằng microgam;
Mbhb là giá trị hiệu chỉnh trắng của kim loại ở nửa sau, tính bằng microgam.
CHÚ THÍCH: Nếu giá trị trắng ở nửa trước (Mfhb) trong khoảng từ 0,0 đến "A" µg ("A" µg bằng 0,2 µg/cm2 trên diện tích thực của cái lọc được dùng.
Mfhb có thể dùng để hiệu chỉnh giá trị Mfh. Nếu Mfhb vượt quá "A" µg thì dùng số lớn hơn trong 2 số sau đây: 1 µg hoặc giá trị nhỏ hơn của Mbhb hoặc 5 % của Mbh.
Nếu giá trị trắng cho nửa sau (Mbhb) ở trong khoảng từ 0,0 đến 1 µg, Mbhb có thể dùng để hiệu chỉnh Mbh. Nếu Mbhb vượt quá 1 µg thì dùng số lớn hơn trong 2 số sau: 1 µg hoặc ít hơn của Mbhb hoặc 5 % của Mbh.
7.5.5 Thủy ngân trong mẫu
7.5.5.1 Phần 1B, nửa trước, Hg. Tính toán lượng thủy ngân thu được ở nửa trước, Phần 1, của hệ thống lấy mẫu theo phương trình:
Hgth = x Vso ln.1 (4)
trong đó:
Hggh là khối lượng tổng của Hg ở nửa trước của hệ thống lấy mẫu (Phần 1), µg;
Qth là lượng Hg trong mẫu phân tích, tính bằng microgam;
Vsoln.1 là thể tích tổng của mẫu được phá (Phần 1), tính bằng mililit;
Vf1B là thể tích Phần 1B đem phân tích, tính bằng mililit. Xem chú thích.
CHÚ THÍCH: Vf1B là thể tích Phần 1B được phân tích. Nếu 1 ml của phần 1B được pha loãng đến 100 ml để phù hợp với khoảng đo thì Vf1B sẽ là 1/100 = 0,01.
7.5.5.2 Các phần 2B, 3A, 3B và 3C, nửa sau, Hg. Tính toán lượng Hg ở Phần 2 bằng phương trình 5 và ở Phần 3A, 3B và 3C bằng phương trình 6. Tính lượng Hg tổng số ở nửa sau của hệ thống lấy mẫu bằng phương trình 7.
Hgbh2 = x Vso ln.2 (5)
trong đó:
Hgbh2 là tổng lượng Hg thu được ở Phần 2, tính bằng microgam;
Qbh2 là lượng Hg trong mẫu phân tích, tính bằng microgam;
Vsoln.2 là thể tích tổng của Phần 2, tính bằng mililit;
Vf2b là thể tích Phần 2B được phân tích, tính bằng mililit (xem chú thích).
CHÚ THÍCH: Vf2b là lượng thực tế của phần 2B được phân tích. Thí dụ, nếu 1 ml của Phần 2B được pha loãng thành 10 ml để đảm bảo nằm trong khoảng phân tích và lấy 5 ml để phân tích, Vf2b sẽ là 0,5. Dùng phương trình 6 để tính lượng Hg ở nửa sau của Phần 3A, 3B và 3C.
Hgbh3(A,B,C) = x Vso ln,3(A, B, C) (6)
trong đó:
Hgbh3 (A, B, C) là tổng lượng Hg thu được ở từng Phần 3A, 3B hoặc 3C, tính bằng microgam;
Qbh(A, B, C) là lượng Hg phân tích được, tính bằng microgam;
Vf3(A, B, C) là thể tích Phần 3A, 3B, 3C đem phân tích, tính bằng mililit. (xem cách tính giống trên);
Vsoln,3(A, B, C) là thể tích tổng Phần 3A, 3B, hoặc 3C, ml.
Hgbh = Hggh2 + Hgbh3A + Hgbh3B + Hbh3C (7)
trong đó:
Hgbh là tổng lượng Hg thu được ở nửa sau của hệ thống lấy mẫu, tính bằng microgam.
7.5.5.3 Tổng lượng Hg trong hệ thống lấy mẫu. Tính lượng tổng số của Hg trong hệ thống lấy mẫu bằng phương trình 8.
Hgt = (Hgfh - Hgfhb) + (Hgbh - Hgbhb) (8)
trong đó:
Hgt là tổng lượng Hg thu được trong hệ thống lấy mẫu, tính bằng microgam;
Hgfhb là giá trị hiệu chỉnh mẫu trắng của Hg thu được ở nửa trước, tính bằng microgam;
Hgbhb là giá trị hiệu chỉnh mẫu trắng của Hg thu được ở nửa sau, tính bằng microgam.
CHÚ THÍCH: Nếu tổng giá trị hiệu chỉnh (Hgfhb + Hgbhb) nằm trong khoảng từ 0 đến 6 µg thì tổng này có thể dùng để hiệu chỉnh mẫu (Hgfh + Hgbh); nếu tổng vượt quá 6 µg thì dùng số lớn hơn trong hai số: 6 µg hoặc 5 % của giá trị mẫu (Hgfg + Hgbh).
7.5.6 Nồng độ kim loại trong khí ống khói. Tính riêng nồng độ kim loại Cd, Cr, As, Ni, Mn, Be, Co, Cu, Pb, P, TI, Ag, Ba, Zn, Se, Sb và Hg trong khí ống khói (không ở điều kiện chuẩn) như sau:
Cs = K4 (Mt / Vm (std))
trong đó:
Cs là nồng độ mỗi kim loại trong khí ống khói, tính bằng miligam trên mét khối;
K4 là 10-3 tính bằng miligam trên microgam;
Mt là tổng lượng mỗi kim loại thu được trong hệ thống lấy mẫu, µg (dùng Hgt, tính toán thay cho Mt);
Vm(std) là thể tích khí khô chuyển về điều kiện chuẩn, tính bằng mét khối.
7.5.7 Biến thiên đẳng tốc và kết quả chấp nhận được. Cũng giống như TCVN 7557-2 : 2005, tính riêng theo điều 6.11 và 6.12.
8 Kiểm soát chất lượng
8.1 Mẫu trắng. Khi phân tích, các mẫu trắng N0 7 đến N0 12 được phá và phân tích như sau: Phá mẫu cái lọc của bình chứa 12 theo 7.2.1, 100 ml của bình chứa N0 7 theo 7.2.2, 100 ml của bình chứa N0 8 theo 7.2.3. Điều đó sinh ra phần mẫu trắng 1A và phần mẫu trắng 1B từ phần mẫu trắng 1. (Có thể phá hai cái lọc riêng rẽ theo 7.2.1, pha loãng riêng đến 300 ml và phân tích riêng, sinh ra hai giá trị trắng cho phần mẫu trắng 1A và phần mẫu trắng 1B. Ba giá trị phần mẫu trắng 1A sẽ được tính như ba giá trị của Mfhb trong phương trình 3 điều 7.5.4.3, sau đó ba giá trị được cộng lại và chia 3 để được giá trị Mfhb dùng để tính Mt theo phương trình 3. Tương tự, ba giá trị của Phần mẫu trắng 1B được tính riêng rẽ, cộng lại, lấy trung bình và được dùng như giá trị Hgfhb trong phương trình 8 điều 7.5.5.3. Phân tích hai cái lọc khác là tùy chọn và phương pháp này không yêu cầu nhưng nếu phân tích thì kết quả được xem như ở trên). Kết hợp 100 ml của bình chứa 8A với 200 ml của bình chứa 9 rồi phá theo 7.2.4. Điều đó sinh ra phần mẫu trắng 2A và phần mẫu trắng 2B từ phần mẫu trắng 2. 100ml của bình chứa N0 8A là phần mẫu trắng 3A. Kết hợp 100ml của bình chứa N010 với 33ml của bình chứa 8B để được phần mẫu trắng 3B. (Dùng 400 ml phần mẫu trắng 3B khi tính giá trị trắng. Dùng thể tích hiện tại khi tính toán các giá trị trắng khác). Pha loãng 225 ml của bình chứa 11 đến 500 ml bằng nước được phần mẫu trắng 3C. Phân tích phần mẫu trắng 1A và phần mẫu trắng 2A theo 7.4.5 hoặc 7.4.6. Phân tích phần mẫu trắng 1B, phần mẫu trắng 2B và phần mẫu trắng 3A, 3B, 3C theo 7.4.7. Phân tích 1A cho giá trị hiệu chỉnh trắng nửa trước của hệ thống, trừ Hg, phân tích phần mẫu trắng 1B cho giá trị hiệu chỉnh trắng cho Hg ở nửa trước của hệ thống. Phân tích phần mẫu trắng 2A cho giá trị hiệu chỉnh cho các kim loại từ Hg của nửa sau hệ thống. Phân tích riêng phần mẫu trắng 2B và 3 cho ta giá trị hiệu chỉnh cho Hg ở nửa sau của hệ thống.
8.2 Mẫu kiểm soát chất lượng. Các mẫu sau dùng để phân tích kiểm soát chất lượng.
8.2.1 Phân tích AA hút trực tiếp hoặc dùng cuvet graphit Sb, As, Ba, Be, Cd, Cr, Co, Cu, Pb, Ni, Mn, Hg, P, Se, Ag, Tl và Zn. Tất cả các mẫu đều được phân tích kép. Phân tích lại một mẫu bất kỳ từ phá mẫu của nửa trước và một của nửa sau. Nếu kết quả nằm ngoài khoảng 75 % và 125 % thì cần phân tích từng mẫu bằng phương pháp thêm chuẩn.
8.2.2 Phân tích Hg bằng AA dùng kỹ thuật hóa hơi lạnh. Tất cả các mẫu đều phân tích kép. Phân tích lại một mẫu của bình axit nitric kể từ công đoạn phá dùng phương pháp đường chuẩn. Kết quả phải nằm trong khoảng 25 %, nếu không thì các mẫu cần được làm theo phương pháp thêm chuẩn.
9 Hiệu quả của phương pháp
9.1 Để đảm bảo độ nhạy các phép đo, nồng độ kim loại nên để cao hơn ít nhất 10 lần giới hạn phát hiện. Trong một số trường hợp và khi hết sức chú ý trong phương pháp phân tích nồng độ đó có thể khoảng ba lần giới hạn phát hiện. Trong mọi trường hợp nên phân tích ít nhất một mẫu kép cho các kim loại, dùng phương pháp thêm (MSA), pha loãng hàng loạt, hoặc thêm chất nền để đảm bảo chất lượng các số liệu.
9.2 Phương pháp xác định giới hạn phát hiện dựa trên các thông số nguồn mẫu và các kết quả phân tích. Nếu cần thì giới hạn phát hiện có thể nhạy hơn là chỉ ra ở Bảng 2 bằng cách:
Lấy mẫu khí 1 h với thể tích khoảng 1,25 m3. Nếu thời gian tăng và lấy đến 5 m3 thì giới hạn phát hiện sẽ là 1/4 giá trị của Bảng 2 (nghĩa là phương pháp nhạy hơn 4 lần so với lấy mẫu 1 h).
Giới hạn phát hiện được làm với giả thiết là tất cả các mẫu đều được phá (trừ phần hút của Hg) và thể tích chất lỏng cuối cùng là 300 ml đối với nửa trước (phần 1) và 150 ml với nửa sau (phần 2A). Nếu thể tích của nửa trước được cô từ 300 ml xuống còn 30 ml, giới hạn phát hiện sẽ là 1/10 của giá trị trên (nhạy hơn 10 lần). Nếu thể tích nửa sau cô từ 150 ml xuống còn 25 ml, giới hạn phát hiện là 1/6 giá trị trên. Hiệu ứng nền rất quan trọng nhất là khi mẫu được cô đặc hơn bình thường. Giảm thể tích xuống dưới 25 ml làm khô cho việc hòa tan cặn và tăng cản trở do các hợp chất khác.
Khi cả hai cải tiến trên được áp dụng đồng thời cho một mẫu thì kết quả cải tiến được nhân lên. Thí dụ, khi thể tích mẫu tăng 5 lần và thể tích phá mẫu của nửa trước và nửa sau giảm 6 lần thì giới hạn phát hiện sẽ giảm 30 lần (phương pháp nhạy hơn 30 lần).
Ngược lại, giảm thể tích mẫu khí và tăng thể tích chất lỏng sẽ làm tăng giới hạn phát hiện (nghĩa là phương pháp sẽ kém nhạy hơn). Mẫu nửa trước và nửa sau (phần 1A và 2A) có thể kết hợp theo tỷ lệ trước khi phân tích. Ghi thể tích chất lỏng tạo ra (trừ của Hg, nó cần được phân tích riêng). Kết hợp mẫu như vậy không cho phép xác định (kể cả nửa trước và nửa sau) mẫu được lấy ở đâu trong hệ thống.
Giới hạn phát hiện như vậy trở nên giá trị đơn cho các kim loại trừ Hg, tuy vẫn phải chú ý tới phần đóng góp của Hg.
Những thảo luận trên không bàn đến hiệu chỉnh trắng.
Bảng 2 - Giới hạn phát hiện (µg/m3) đối với phần mẫu của hệ thống lấy mẫu sử dụng phương pháp ICP-AES và AAS
|
Kim loại |
Nửa trước của hệ thống lấy mẫu Phần 1 Đầu lấy mẫu & cái lọc |
Nửa sau của hệ thống lấy mẫu Phần 2 Bình hấp thu (impinger) 1 - 3 |
Nửa sau của hệ thống lấy mẫu Phần 3 Bình hấp thu (impinger) 4 - 5 |
Toàn bộ hệ thống lấy mẫu |
|
Antimoan/Stibi (Sb) |
7,7 (0,7) * |
3,8 (0,4) * |
|
11,5 (1,1) * |
|
Asen (As) |
12,7 (0,3) * |
6,4 (0,1) * |
|
19,1 (0,4) * |
|
Bari (Ba) |
0,5 |
0,3 |
|
0,8 |
|
Beri (Be) |
0,07 (0,05) * |
0,04 (0,03) * |
|
0,11 (0,08) * |
|
Cadmi (Cd) |
1,0 (0,02) * |
0,5 (0,01) * |
|
1,5 (0,03) * |
|
Crom (Cr) tổng |
1,7 (0,2) * |
0,8 (0,1) * |
|
2,5 (0,3) * |
|
Coban (Co) |
1,7 (0,2) * |
0,8 (0,1) * |
|
2,5 (0,3) * |
|
Đồng (Cu) |
1,4 |
0,7 |
|
2,1 |
|
Chì (Pb) |
10,1 (0,2) * |
5,0 (0,1) * |
|
15,1 (0,3) * |
|
Mangan (Mn) |
0,5 (0,2) * |
0,2 (0,1) * |
|
0,7 (0,3) * |
|
Thủy ngân (Hg) |
0,6** |
3,0** |
2,0** |
5,6** |
|
Niken (Ni) |
3,6 |
1,8 |
|
5,4 |
|
Phospho (P) |
18,0 |
9,0 |
|
27,0 |
|
Selen (Se) |
18,0 (0,5) * |
9,0 (0,3) * |
|
27 (0,8) * |
|
Bạc (Ag) |
1,7 |
0,9 |
|
2,6 |
|
Tali (TI) |
9,6 (0,2) * |
4,8 (0,1) * |
|
14,4 (0,3) * |
|
Kẽm (Zn) |
0,5 |
0,3 |
|
0,8 |
CHÚ THÍCH 1:
()*: Giới hạn phát hiện khi phân tích bằng GF-AAS.
**: Giới hạn phát hiện khi phân tích bằng CV- AAS, ước tính đối với nửa sau và toàn bộ hệ thống lấy mẫu.
CHÚ THÍCH 2:
Thông thường, giới hạn phát hiện được xác định dựa vào các thông số thực tế và kết quả phân tích được mô tả ở điều bên trên điều này.
9.3 Dùng (1) cách tiến hành của phương pháp, (2) giới hạn phát hiện trình bày ở điều 1, (3) thể tích 300 ml với nửa trước và 150 ml với nửa sau và (4) mẫu khí 1,25 m3, giới hạn phát hiện như trên Bảng 2, được tính như sau:
= D
trong đó:
A là giới hạn phát hiện phân tích, tính bằng microgam trên mililit;
B là thể tích mẫu dùng cho phân tích, tính bằng mililit;
C là thể tích mẫu khí, dscm (dsm3);
D là giới hạn phát hiện trong ống khói, tính bằng microgam trên mét khối.
Giá trị ở Bảng 2 được tính cho nửa trước, nửa sau và toàn bộ hệ thống.

Hình 2 - Sơ đồ thu mẫu và xử lý mẫu
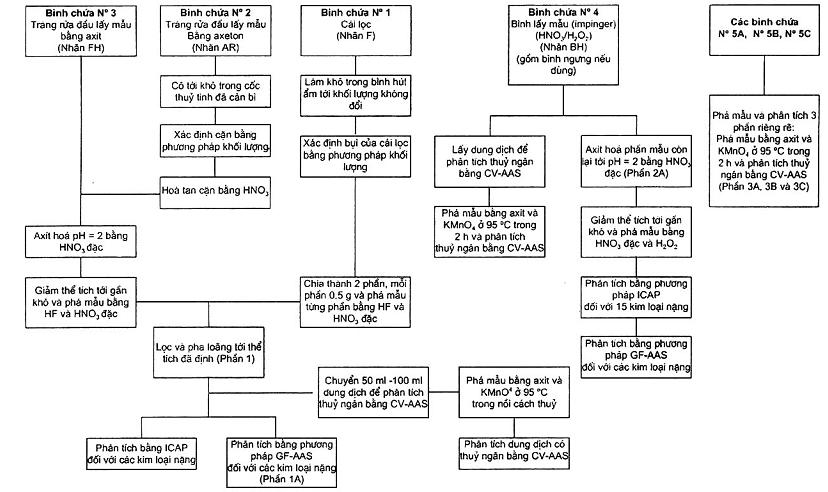
Hình 3 - Sơ đồ chuẩn bị mẫu và phân tích mẫu
Phụ lục A
(Tham khảo)
Thư mục tài liệu tham khảo
1. Method 303F, Standard methods for the examination of water and wastewater, available from American Public Health Association, 1015 18 th Street, NW, Washington, D.C 20036.
2. EPA Method 200.7 Code of Federal Regulation. Title 40, Part 136, Appendix C.
3. EPA Methods 1 though 5. 12, and 29 Code of Federal Regulation. Title 40, Part 60, Appendix A.
4. ASTM Standard methods D2986 - 71, available from American Sociaty for testing and Materianls, 1916 Race Street, Philadelphia, PA 19103.
1) Nếu Phần 1A và 2A nhập lại cần lấy phần hút tỷ lệ. Phương trình 1-3 phản ánh điều này.


