Tiêu chuẩn Việt Nam TCVN ISO 13485:2004 về Dụng cụ y tế - Hệ thống quản lý chất lượng - Yêu cầu đối với các mục đích chế định do Bộ Khoa học và Công nghệ ban hành đã được thay thế bởi Tiêu chuẩn quốc gia TCVN ISO 13485:2017 (ISO 13485:2016) về Trang thiết bị y tế - Hệ thống quản lý chất lượng - Yêu cầu đối với các mục đích chế định .
Nội dung toàn văn Tiêu chuẩn Việt Nam TCVN ISO 13485:2004 về Dụng cụ y tế - Hệ thống quản lý chất lượng - Yêu cầu đối với các mục đích chế định do Bộ Khoa học và Công nghệ ban hành
TIÊU CHUẨN VIỆT NAM
TCVN ISO 13485 : 2004
ISO 13485 : 2003
DỤNG CỤ Y TẾ – HỆ THỐNG QUẢN LÝ CHẤT LƯỢNG – YÊU CẦU ĐỐI VỚI CÁC MỤC ĐÍCH CHẾ ĐỊNH
Medical devices – Quality management systems – Requirements for regulatory purposes
Mục lục
Lời nói đầu
Lời giới thiệu
0.1. Khái quát
0.2. Tiếp cận theo quá trình
0.3. Mối tương quan với các tiêu chuẩn khác
0.4. Sự tương hợp với các hệ thống quản lý khác
1. Phạm vi áp dụng
1.1. Khái quát
1.2. Áp dụng
2. Tài liệu viện dẫn
3. Thuật ngữ và định nghĩa
4. Hệ thống quản lý chất lượng
4.1. Các yêu cầu chung
4.2. Yêu cầu về hệ thống tài liệu
5. Trách nhiệm của lãnh đạo
5.1. Cam kết của lãnh đạo
5.2. Hướng vào khách hàng
5.3 Chính sách chất lượng
5.4. Hoạch định
5.5. Trách nhiệm, quyền hạn và trao đổi thông
5.6. Xem xét của lãnh đạo
6. Quản lý nguồn lực
6.1. Cung cấp nguồn lực
6.2. Nguồn nhân lực
6.3. Cơ sở làm việc
6.4. Môi trường làm việc
7. Tạo sản phẩm
7.1. Hoạch định việc tạo sản phẩm
7.2. Các quá trình liên quan đến khách hàng
7.3. Thiết kế và phát triển
7.4. Mua hàng
7.5. Sản xuất và cung cấp dịch vụ
7.6 Kiểm soát phương tiện theo dõi và đo lường
8 Đo lường, phân tích và cải tiến
8.1 Khái quát
8.2. Theo dõi và đo lường
8.3. Kiểm soát sản phẩm không phù hợp
8.4. Phân tích dữ liệu
8.5. Cải tiến
Phụ lục A (tham khảo) Tương ứng giữa TCVN ISO 13485:2004 và ISO 13485:1996
Phụ lục B (tham khảo) Giải thích về những khác biệt giữa TCVN ISO 13485:2004 và TCVN ISO 9001:2000
Thư mục tài liệu tham khảo
Lời nói đầu
TCVN ISO 13485 : 2004 hoàn toàn tương đương với ISO13485 : 2003.
TCVN ISO 13485 : 2004 do Ban kỹ thuật tiêu chuẩn TCVN/TC 176, Quản lý chất lượng và đảm bảo chất lượng biên soạn, Tổng cục Tiêu chuẩn Đo lường Chất lượng đề nghị, Bộ Khoa học và Công nghệ ban hành.
Lời giới thiệu
0.1. Khái quát
Tiêu chuẩn này quy định các yêu cầu đối với hệ thống quản lý chất lượng có thể sử dụng bởi tổ chức thiết kế, phát triển, sản xuất, lắp đặt và bảo dưỡng dụng cụ y tế và bởi tổ chức thiết kế, phát triển và cung cấp dịch vụ liên quan.
Tiêu chuẩn này còn được các tổ chức nội bộ và bên ngoài, bao gồm các tổ chức chứng nhận, sử dụng để đánh giá khả năng của tổ chức đó đối với việc đáp ứng những yêu cầu chế định và yêu cầu của khách hàng.
Thông tin ở "CHÚ THÍCH" là để giải thích hoặc làm rõ các yêu cầu.
Lưu ý rằng các yêu cầu đối với hệ thống quản lý chất lượng quy định trong tiêu chuẩn này là để bổ sung cho những yêu cầu kỹ thuật đối với sản phẩm.
Việc chấp nhận hệ thống quản lý chất lượng cần là một quyết định chiến lược của tổ chức. Việc thiết kế và áp dụng hệ thống quản lý chất lượng của một tổ chức phụ thuộc vào các nhu cầu khác nhau, các mục tiêu riêng biệt, các sản phẩm cung cấp, các quá trình được sử dụng, quy mô và cơ cấu của tổ chức đó. Tiêu chuẩn này không hướng tới sự đồng nhất về cấu trúc của các hệ thống quản lý chất lượng hoặc của sự đồng nhất của hệ thống tài liệu.
Có rất nhiều chủng loại dụng cụ y tế và một số yêu cầu của tiêu chuẩn này chỉ áp dụng cho các nhóm dụng cụ y tế được nêu ra. Các nhóm này được xác định ở Điều 3.
0.2. Tiếp cận theo quá trình
Tiêu chuẩn này dựa vào cách tiếp cận theo quá trình đối với quản lý chất lượng.
Mọi hoạt động tiếp nhận đầu vào và chuyển hóa chúng thành đầu ra đều có thể được xem là quá trình.
Để tổ chức hoạt động có hiệu lực, cần xác định và quản lý hàng loạt quá trình liên kết với nhau.
Đầu ra của một quá trình thường chính là đầu vào của quá trình tiếp sau.
Việc áp dụng hệ thống các quá trình trong một tổ chức, cùng với sự nhận biết các mối tương tác giữa các quá trình như vậy, và việc quản lý chúng, có thể được coi là "cách tiếp cận theo quá trình".
0.3. Mối quan hệ với các tiêu chuẩn khác
0.3.1. Mối quan hệ với TCVN ISO 9001
Khi là tiêu chuẩn độc lập, tiêu chuẩn này dựa vào TCVN ISO 9001.
Các điều hoặc quy định được trích dẫn trực tiếp và không thay đổi so với TCVN ISO 9001 được thể hiện bằng kiểu chữ đứng. Phụ lục B trình bày những điều và quy định không thay đổi này.
Khi mà nội dung quy định của tiêu chuẩn này không tương đương với nội dung quy định của TCVN ISO 9001 thì toàn bộ câu hoặc đoạn có phần nội dung quy định đó được thể hiện bằng kiểu chữ in nghiêng (nếu là bản điện tử thì sử dụng chữ in nghiêng màu xanh). Bản chất của những thay đổi trong nội dung quy định và nguyên nhân dẫn tới chúng được nêu ở Phụ lục B.
0.3.2. Mối quan hệ với ISO/TR 14969
ISO/TR 14969 là báo cáo kỹ thuật đưa ra những hướng dẫn về áp dụng ISO 13485.
0.4. Sự tương hợp với các hệ thống quản lý khác
Tiêu chuẩn này được trình bày theo dạng thức trình bày của TCVN ISO 9001 để tạo thuận tiện cho người sử dụng tiêu chuẩn trong cộng đồng những người sử dụng dụng cụ y tế.
TIêu chuẩn này không quy định các yêu cầu riêng đối với những hệ thống quản lý khác, chẳng hạn những yêu cầu cụ thể đối với hệ thống quản lý môi trường, hệ thống quản lý sức khỏe nghề nghiệp và an toàn hoặc hệ thống quản lý tài chính.
Tuy nhiên, tiêu chuẩn này tạo điều kiện cho tổ chức tiệm cận hoặc kết hợp hệ thống quản lý chất lượng của mình với các yêu cầu đối với các hệ thống quản lý liên quan. Điều này giúp cho tổ chức có khả năng chuyển đổi (các) hệ thống quản lý hiện hành của mình nhằm thiết lập hệ thống quản lý chất lượng phù hợp với yêu cầu của tiêu chuẩn này.
TCVN ISO 13485 : 2004
DỤNG CỤ Y TẾ – HỆ THỐNG QUẢN LÝ CHẤT LƯỢNG – YÊU CẦU ĐỐI VỚI CÁC MỤC ĐÍCH CHẾ ĐỊNH
Medical devices – Quality management systems – Requirements for regulatory purposes
1. Phạm vi áp dụng
1.1. Khái quát
Tiêu chuẩn này quy định các yêu cầu đối với hệ thống quản lý chất lượng trong đó một tổ chức cần thể hiện khả năng cung cấp dụng cụ y tế và dịch vụ liên quan đáp ứng đúng những yêu cầu của khách hàng và yêu cầu chế định áp dụng cho dụng cụ y tế và dịch vụ liên quan.
Mục tiêu hàng đầu của tiêu chuẩn này là thúc đẩy việc áp dụng các yêu cầu chế định hài hòa về dụng cụ y tế đối với hệ thống quản lý chất lượng. Vì vậy, tiêu chuẩn này bao gồm một số yêu cầu cụ thể đối với dụng cụ y tế và không bao gồm một số yêu cầu của TCVN ISO 9001 không phù hợp làm yêu cầu chế định. Do những ngoại lệ này, các tổ chức có hệ thống quản lý chất lượng phù hợp với tiêu chuẩn này không thể công bố sự phù hợp của chúng với TCVN ISO 9001 trừ khi chúng phù hợp với tất cả các yêu cầu của TCVN ISO 9001 (xem Phụ lục B).
1.2. Áp dụng
Tất cả các yêu cầu của tiêu chuẩn này đều là những yêu cầu áp dụng riêng cho những tổ chức cung cấp dụng cụ y tế, bất kể những tổ chức này thuộc loại hình nào hoặc có quy mô như thế nào.
Nếu các yêu cầu chế định cho phép có các ngoại lệ về kiểm soát thiết kế và phát triển (xem 7.3) thì điều này có thể sử dụng để giải thích cho việc đưa những ngoại lệ đó vào hệ thống quản lý chất lượng. Các văn bản pháp quy này có thể cung cấp những thỏa thuận khác sẽ được đề cập đến trong hệ thống quản lý chất lượng. Trách nhiệm của tổ chức là đảm bảo rằng những công bố về sự phù hợp với tiêu chuẩn này sẽ phản ánh sự ngoại lệ đối với kiểm soát thiết kế và phát triển [xem 4.2.2 a) và 7.3].
Nếu mọi yêu cầu trong Điều 7 của tiêu chuẩn này đều không áp dụng được do dụng cụ y tế là đối tượng áp dụng của hệ thống quản lý chất lượng thì tổ chức không cần thiết phải đưa những yêu cầu đó vào hệ thống quản lý chất lượng của mình [xem 4.2.2 a)].
Các quá trình mà tiêu chuẩn này yêu cầu và áp dụng được cho dụng cụ y tế nhưng không được tổ chức thực hiện thì điều này thuộc trách nhiệm của tổ chức và chúng đều phải được giải thích trong hệ thống quản lý chất lượng của tổ chức [xem 4.1 a)].
Trong tiêu chuẩn này, các cụm từ "nếu thích hợp" và "ở nơi thích hợp" được sử dụng nhiều lần. Khi một yêu cầu được bổ nghĩa bởi các cụm từ này thì yêu cầu đó dường như là "thích hợp" trừ khi tổ chức có thể nêu trong tài liệu cách giải thích khác. Một yêu cầu được xem là "thích hợp" nếu đó là yêu cầu cần thiết để:
- Sản phẩm đáp ứng các yêu cầu quy định;
- Tổ chức tiến hành hành động khắc phục.
2. Tiêu chuẩn viện dẫn
TCVN ISO 9000 : 2000 (ISO 9000 : 2000) Hệ thống quản lý chất lượng − Cơ sở và từ vựng.
3. Thuật ngữ và định nghĩa
Tiêu chuẩn này áp dụng các thuật ngữ và định nghĩa cho trong TCVN ISO 9000 cùng với các thuật ngữ, định nghĩa dưới đây.
Các thuật ngữ dưới đây, sử dụng trong tiêu chuẩn này để mô tả chuỗi cung ứng, đã được thay đổi để phản ánh từ vựng hiện hành:
Người cung ứng → Tổ chức → Khách hàng
Thuật ngữ "tổ chức" thay thế cho thuật ngữ "người cung ứng" đã sử dụng trong ISO 13485:1996 và chỉ đơn vị áp dụng tiêu chuẩn này. Hiện nay, thuật ngữ "người cung ứng" còn thay thế cho thuật ngữ "người thầu phụ".
Trong tiêu chuẩn này, thuật ngữ "sản phẩm" cũng có nghĩa là "dịch vụ".
Khi mà các yêu cầu được quy định để áp dụng cho "dụng cụ y tế" thì chúng cũng áp dụng đầy đủ cho những dịch vụ liên quan do tổ chức cung cấp.
Các thuật ngữ dưới đây cần được coi là những thuật ngữ chung do các định nghĩa nêu trong những văn bản pháp quy quốc gia có thể khác biệt đôi chút và được ưu tiên hơn.
3.1. Dụng cụ y tế cấy ghép hoạt tính (active implantable medical device)
Dụng cụ y tế hoạt tính được trù định đưa từng phần hoặc toàn bộ vào cơ thể con người bằng phẫu thuật hoặc bằng biện pháp y học hoặc vào vị trí tự nhiên ở bên ngoài cơ thể bằng can thiệp y học và được trù định duy trì tại đó sau khi cấy ghép.
3.2. Dụng cụ y tế hoạt tính (active medical device)
Dụng cụ y tế hoạt động dựa vào nguồn điện năng hoặc nguồn năng lượng bất kỳ không phải được tạo ra trực tiếp bởi cơ thể con người hoặc bởi trọng lực.
3.3. Thông báo tư vấn (advisory notice)
Thông báo do tổ chức cung ứng dụng cụ y tế đưa ra tiếp sau khi giao hàng để cung cấp các thông tin bổ sung và/hoặc tư vấn về việc cần phải thực hiện hành động cần thiết nào trong:
- Sử dụng dụng cụ y tế;
- Cải biến dụng cụ y tế;
- Hoàn trả dụng cụ y tế cho tổ chức cung ứng;
- Phá hủy dụng cụ y tế.
CHÚ THÍCH: Việc đưa ra một thông báo tư vấn có thể được yêu cầu để phù hợp với văn bản pháp quy quốc gia hoặc khu vực.
3.4. Khiếu nại của khách hàng (customer complaint)
Thông tin trao đổi bằng văn bản, bằng dạng thức điện tử hoặc bằng miệng thông báo về những khuyết, nhược điểm liên quan đến tính đồng nhất, chất lượng, tuổi thọ, độ tin cậy, an toàn hoặc tính năng của dụng cụ y tế đã được đưa ra thị trường.
3.5. Dụng cụ y tế cấy ghép (implantable medical device)
Dụng cụ y tế được trù định để:
- Đưa toàn bộ hoặc từng phần vào cơ thể con người hoặc vào vị trí tự nhiên ở bên ngoài cơ thể;
- Thay thế cho bề mặt biểu mô hoặc bề mặt của mắt.
Bằng biện pháp can thiệp phẫu thuật và được trù định giữ lại sau khi cấy ghép ít nhất 30 ngày và sẽ chỉ được tháo ra cũng bằng can thiệp y học hoặc phẫu thuật.
CHÚ THÍCH: Định nghĩa này áp dụng cho các dụng cụ y tế cấy ghép không phải là dụng cụ y tế cấy ghép hoạt tính.
3.6. Thông tin ghi nhãn (labelling)
Thông tin bằng văn bản, bằng dạng thức in ấn hoặc đồ hoạ
- Được gắn vào dụng cụ y tế hoặc đồ chứa hay bao gói của dụng cụ này;
hoặc:
- Kèm theo dụng cụ y tế
liên quan đến việc nhận biết, mô tả kỹ thuật và sử dụng dụng cụ đó nhưng ngoại trừ tài liệu giao nhận.
CHÚ THÍCH: Một số văn bản pháp quy quốc gia và khu vực đề cập tới "thông tin ghi nhãn" như là "thông tin do nhà sản xuất cung cấp".
3.7. Dụng cụ y tế (medical device)
Bất kỳ dụng cụ, thiết bị, công cụ, khí cụ, phần cấy ghép, chất thử nhân tạo hoặc dụng cụ đánh dấu, phần mềm, vật liệu hoặc hạng mục tương tự hay liên quan nào khác mà nhà sản xuất trù định sẽ sử dụng riêng biệt hoặc kết hợp cho con người với một hoặc nhiều mục đích cụ thể của việc:
- Chẩn đoán, phòng ngừa, theo dõi, điều trị hoặc làm giảm bệnh tật;
- Chẩn đoán, theo dõi, điều trị, làm giảm hoặc đắp vào vết thương;
- Khám, thay thế, cải biến hoặc hỗ trợ cho việc giải phẫu hoặc quá trình sinh lý;
- Hỗ trợ việc duy trì cuộc sống;
- Kiểm soát sự thụ thai;
- Tẩy trùng dụng cụ y tế;
- Cung cấp thông tin cho mục đích y học bằng biện pháp kiểm tra mẫu phẩm lấy ra từ cơ thể con người;
và không đạt được hành động trù định ban đầu trong hoặc trên cơ thể con người bằng biện pháp dược lý, miễn dịch hoặc trao đổi chất nhưng có thể được trợ giúp trong quá trình sử dụng bằng những biện pháp như vậy.
CHÚ THÍCH: Định nghĩa này đã được phát triển bởi Tổ công tác đặc trách về hài hòa toàn cầu (GHTF). Xem thư mục tài liệu tham khảo [15].
3.8. Dụng cụ y tế vô trùng (sterile medical device)
Chủng loại dụng cụ y tế trù định để đáp ứng các yêu cầu vô trùng.
CHÚ THÍCH: Các yêu cầu vô trùng dụng cụ y tế có thể là đối tượng của văn bản pháp quy hoặc tiêu chuẩn quốc gia hay khu vực.
4. Hệ thống quản lý chất lượng
4.1. Yêu cầu chung
Tổ chức phải xây dựng, lập văn bản, thực hiện, duy trì hệ thống quản lý chất lượng và duy trì hiệu lực của hệ thống theo các yêu cầu của tiêu chuẩn này.
Tổ chức phải:
a) nhận biết các quá trình cần thiết trong hệ thống quản lý chất lượng và áp dụng chúng trong toàn bộ tổ chức (xem 1.2);
b) xác định trình tự và mối liên hệ của các quá trình;
c) xác định chuẩn mực và phương pháp cần thiết để đảm bảo việc tác nghiệp và kiểm soát các quá trình có hiệu lực;
d) đảm bảo sự sẵn có của các nguồn lực và thông tin cần thiết để hỗ trợ hoạt động tác nghiệp và theo dõi các quá trình này;
e) đo lường, theo dõi và phân tích các quá trình;
f) thực hiện các hành động cần thiết để đạt được kết quả dự định và duy trì hiệu lực của các quá trình này.
Tổ chức phải quản lý các quá trình tuân thủ theo các yêu cầu của tiêu chuẩn.
Khi tổ chức chọn nguồn bên ngoài cho bất kỳ quá trình nào ảnh hưởng đến sự phù hợp của sản phẩm với các yêu cầu, tổ chức phải đảm bảo kiểm soát được những quá trình đó. Việc kiểm soát những quá trình do nguồn bên ngoài phải được nhận biết trong hệ thống quản lý chất lượng (xem 8.5.1).
CHÚ THÍCH: Các quá trình cần thiết đối với hệ thống quản lý chất lượng nêu trên cần bao gồm cả các quá trình hoạt động quản lý, cung cấp nguồn lực, tạo sản phẩm và đo lường.
4.2. Yêu cầu về hệ thống tài liệu
4.2.1. Khái quát
Các tài liệu của hệ thống quản lý chất lượng phải bao gồm:
a) văn bản công bố về chính sách chất lượng và mục tiêu chất lượng;
b) sổ tay chất lượng;
c) các thủ tục dạng văn bản theo yêu cầu của tiêu chuẩn này;
d) các tài liệu cần có của tổ chức để đảm bảo việc hoạch định, tác nghiệp và kiểm soát có hiệu lực các quá trình của tổ chức đó;
e) các hồ sơ theo yêu cầu của tiêu chuẩn này (xem 4.2.4).
f) mọi tài liệu khác được quy định bởi các văn bản pháp quy quốc gia hoặc khu vực.
Ở trong tiêu chuẩn này, nếu như có quy định rằng một yêu cầu, thủ tục, hoạt động hoặc thỏa thuận đặc biệt được "lập thành văn bản" thì yêu cầu, thủ tục, hoạt động hoặc thỏa thuận đặc biệt đó phải được áp dụng và duy trì.
Đối với mỗi loại hoặc kiểu dụng cụ y tế, tổ chức phải thiết lập và duy trì một tệp tài liệu gồm có hoặc xác định các tài liệu định rõ những quy định đối với sản phẩm và yêu cầu đối với hệ thống quản lý chất lượng (xem 4.2.3). Các tài liệu này định rõ quá trình chế tạo hoàn chỉnh và, nếu thích hợp, lắp đặt và dịch vụ bảo dưỡng.
CHÚ THÍCH 1: Mức độ văn bản hóa hệ thống quản lý chất lượng của mỗi tổ chức có thể khác nhau tuỳ thuộc vào:
a) quy mô của tổ chức và loại hình hoạt động;
b) sự phức tạp và sự liên hệ giữa các quá trình;
c) năng lực của con người.
CHÚ THÍCH 2: Hệ thống tài liệu được thể hiện dưới bất kỳ dạng hoặc loại phương tiện truyền thông nào.
4.2.2. Sổ tay chất lượng
Tổ chức phải lập và duy trì sổ tay chất lượng trong đó bao gồm:
a) phạm vi của hệ thống quản lý chất lượng, bao gồm cả các nội dung chi tiết và sự giải thích về mọi ngoại lệ và/hoặc những quy định không áp dụng (xem 1.2);
b) các thủ tục dạng văn bản được thiết lập cho hệ thống quản lý chất lượng hoặc viện dẫn đến chúng;
c) mô tả sự tương tác giữa các quá trình trong hệ thống quản lý chất lượng.
Sổ tay chất lượng cần nêu rõ cấu trúc của hệ thống tài liệu được sử dụng trong hệ thống quản lý chất lượng.
4.2.3. Kiểm soát tài liệu
Các tài liệu theo yêu cầu của hệ thống quản lý chất lượng phải được kiểm soát. Hồ sơ chất lượng là một loại tàì liệu đặc biệt và phải được kiểm soát theo các yêu cầu nêu trong 4.2.4 .
Phải lập một thủ tục dạng văn bản để xác định việc kiểm soát cần thiết nhằm:
a) xem xét và phê duyệt tài liệu về sự thỏa đáng trước khi ban hành;
b) xem xét, cập nhật khi cần và phê duyệt lại tài liệu;
c) đảm bảo nhận biết được các thay đổi và tình trạng sửa đổi hiện hành của tài liệu;
d) đảm bảo các bản của các tài liệu thích hợp sẵn có ở nơi sử dụng;
e) đảm bảo tài liệu luôn rõ ràng, dễ nhận biết;
f) đảm bảo các tài liệu có nguồn gốc bên ngoài được nhận biết và việc phân phối chúng được kiểm soát;
g) ngăn ngừa việc sử dụng vô tình các tài liệu lỗi thời và áp dụng các dấu hiệu nhận biết thích hợp nếu chúng được giữ lại vì mục đích nào đó.
Tổ chức phải đảm bảo rằng những thay đổi trong tài liệu đều được xem xét và phê duyệt bởi những người đã phê duyệt tài liệu trước đó hoặc bởi những người được chỉ định khác có khả năng tiếp cận được với thông tin gốc thích hợp làm cơ sở cho việc đưa ra quyết định.
Tổ chức phải xác định thời hạn lưu giữ đối với các tài liệu được kiểm soát đã lỗi thời, ít nhất một bản đối với từng tài liệu. Thời hạn này phải đảm bảo được rằng những tài liệu mà theo đó các dụng cụ y tế đã được chế tạo và thử nghiệm luôn sẵn có ít nhất trong thời gian sử dụng của dụng cụ y tế liên quan do tổ chức xác định nhưng không ngắn hơn thời hạn lưu giữ của bất kỳ hồ sơ nào (xem 4.2.4) hoặc thời hạn đã được quy định bởi yêu cầu chế định liên quan.
4.2.4. Kiểm soát hồ sơ
Phải lập và duy trì các hồ sơ để cung cấp bằng chứng về sự phù hợp với các yêu cầu và hoạt động tác nghiệp có hiệu lực của hệ thống quản lý chất lượng. Các hồ sơ chất lượng phải rõ ràng, dễ nhận biết và dễ sử dụng. Phải lập một thủ tục bằng văn bản để xác định việc kiểm soát cần thiết đối với việc nhận biết, bảo quản, bảo vệ, sử dụng, xác định thời gian lưu giữ và hủy bỏ các hồ sơ chất lượng.
Tổ chức phải lưu giữ các hồ sơ trong thời hạn ít nhất là bằng thời hạn sử dụng của dụng cụ y tế mà tổ chức đã xác định nhưng không ngắn hơn thời hạn 2 năm kể từ ngày sản phẩm đó được đưa ra thị trường hoặc thời hạn đã được quy định bởi yêu cầu chế định liên quan.
5. Trách nhiệm của lãnh đạo
5.1. Cam kết của lãnh đạo
Lãnh đạo cao nhất phải cung cấp bằng chứng về sự cam kết của mình đối với việc xây dựng và thực hiện hệ thống quản lý chất lượng và duy trì hiệu lực của hệ thống đó bằng cách:
a) truyền đạt cho tổ chức về tầm quan trọng của việc đáp ứng khách hàng cũng như các yêu cầu của pháp luật và chế định;
b) thiết lập chính sách chất lượng;
c) đảm bảo việc thiết lập các mục tiêu chất lượng;
d) tiến hành việc xem xét của lãnh đạo;
e) đảm bảo sẵn có các nguồn lực.
CHÚ THÍCH: Với mục đích của tiêu chuẩn này, các yêu cầu của pháp luật chỉ giới hạn ở độ an toàn và tính năng sử dụng của dụng cụ y tế.
5.2. Hướng vào khách hàng
Lãnh đạo cao nhất phải đảm bảo rằng các yêu cầu của khách hàng được xác định và đáp ứng (xem 7.2.1 và 8.2.1).
5.3. Chính sách chất lượng
Lãnh đạo cao nhất phải đảm bảo rằng chính sách chất lượng:
a) phù hợp với mục đích của tổ chức;
b) bao gồm việc cam kết đáp ứng các yêu cầu và duy trì hiệu lực của hệ thống quản lý chất lượng;
c) cung cấp cơ sở cho việc thiết lập và xem xét các mục tiêu chất lượng;
d) được truyền đạt và thấu hiểu trong tổ chức;
e) được xem xét để luôn thích hợp.
5.4. Hoạch định
5.4.1. Mục tiêu chất lượng
Lãnh đạo cao nhất phải đảm bảo rằng mục tiêu chất lượng, bao gồm cả những điều cần thiết để đáp ứng các yêu cầu của sản phẩm [xem 7.1 a)], được thiết lập tại mọi cấp và từng bộ phận chức năng thích hợp trong tổ chức. Mục tiêu chất lượng phải đo được và nhất quán với chính sách chất lượng.
5.4.2. Hoạch định hệ thống quản lý chất lượng
Lãnh đạo cao nhất phải đảm bảo
a) tiến hành hoạch định hệ thống quản lý chất lượng để đáp ứng các yêu cầu nêu trong 4.1 cũng như các mục tiêu chất lượng, và
b) tính nhất quán của hệ thống quản lý chất lượng được duy trì khi các thay đổi đối với hệ thống quản lý chất lượng được hoạch định và thực hiện.
5.5. Trách nhiệm, quyền hạn và trao đổi thông tin
5.5.1. Trách nhiệm và quyền hạn
Lãnh đạo cao nhất phải đảm bảo các trách nhiệm, quyền hạn và mối quan hệ của chúng được xác định, được lập thành văn bản và thông báo trong tổ chức.
Lãnh đạo cao nhất phải thiết lập mối quan hệ tương tác giữa tất cả mọi người chịu sự lãnh đạo của mình, thực hiện và kiểm tra xác nhận công việc ảnh hưởng tới chất lượng và phải đảm bảo sự độc lập và quyền hạn cần thiết cho việc thực hiện những nhiệm vụ này.
CHÚ THÍCH: Các văn bản pháp quy quốc gia hoặc khu vực có thể yêu cầu việc chỉ định những người cụ thể chịu trách nhiệm về các hoạt động liên quan đến việc theo dõi những sự kiện diễn ra ở giai đoạn tiền sản xuất và thông báo về những sự việc bất lợi (xem 8.2.1 và 8.5.1).
5.5.2. Đại diện của lãnh đạo
Lãnh đạo cao nhất phải chỉ định một thành viên trong ban lãnh đạo, ngoài các trách nhiệm khác, có trách nhiệm và quyền hạn bao gồm
a) đảm bảo các quá trình cần thiết của hệ thống quản lý chất lượng được thiết lập, thực hiện và duy trì;
b) báo cáo cho lãnh đạo cao nhất về kết quả hoạt động của hệ thống quản lý chất lượng và về mọi nhu cầu cải tiến, và
c) đảm bảo thúc đẩy toàn bộ tổ chức nhận thức được các yêu cầu chế định và các yêu cầu của khách hàng.
CHÚ THÍCH: Trách nhiệm của đại diện lãnh đạo về chất lượng có thể bao gồm cả quan hệ với bên ngoài về các vấn đề có liên quan đến hệ thống quản lý chất lượng.
5.5.3. Trao đổi thông tin nội bộ
Lãnh đạo cao nhất phải đảm bảo thiết lập các quá trình trao đổi thông tin thích hợp trong tổ chức và có sự trao đổi thông tin về hiệu lực của hệ thống quản lý chất lượng.
5.6. Xem xét của lãnh đạo
5.6.1. Khái quát
Lãnh đạo cao nhất phải định kỳ xem xét hệ thống quản lý chất lượng, để đảm bảo nó luôn thích hợp, thỏa đáng, và có hiệu lực. Việc xem xét này phải đánh giá được cơ hội cải tiến và nhu cầu thay đổi đối với hệ thống quản lý chất lượng của tổ chức, kể cả chính sách chất lượng và các mục tiêu chất lượng.
Hồ sơ xem xét của lãnh đạo phải được duy trì (xem 4.2.4)
5.6.2. Đầu vào của việc xem xét
Đầu vào của việc xem xét của lãnh đạo phải bao gồm thông tin về
a) kết quả của các cuộc đánh giá,
b) phản hồi của khách hàng,
c) việc thực hiện các quá trình và sự phù hợp của sản phẩm,
d) tình trạng của các hành động khắc phục và phòng ngừa,
e) các hành động tiếp theo từ các cuộc xem xét của lãnh đạo lần trước,
f) những thay đổi có thể ảnh hưởng đến hệ thống quản lý chất lượng, và
g) các khuyến nghị về cải tiến,
h) các yêu cầu chế định mới hoặc đã được sửa đổi.
5.6.3. Đầu ra của việc xem xét
Đầu ra của việc xem xét của lãnh đạo phải bao gồm mọi quyết định và hành động liên quan đến:
a) các cải tiến cần thiết để duy trì tính hiệu lực của hệ thống quản lý chất lượng và các quá trình của hệ thống;
b) việc cải tiến các sản phẩm liên quan đến yêu cầu của khách hàng;
c) nhu cầu về nguồn lực.
6. Quản lý nguồn lực
6.1. Cung cấp nguồn lực
Tổ chức phải xác định và cung cấp các nguồn lực cần thiết để:
a) thực hiện và duy trì hệ thống quản lý chất lượng và duy trì hiệu lực của hệ thống đó;
b) đáp ứng các yêu cầu chế định và yêu cầu của khách hàng.
6.2. Nguồn nhân lực
6.2.1. Khái quát
Những người thực hiện các công việc ảnh hưởng đến chất lượng sản phẩm phải có năng lực trên cơ sở được giáo dục, đào tạo, có kỹ năng và kinh nghiệm thích hợp.
6.2.2. Năng lực, nhận thức và đào tạo
Tổ chức phải
a) xác định năng lực cần thiết của những người thực hiện các công việc ảnh hưởng đến chất lượng sản phẩm,
b) tiến hành đào tạo hay những hành động khác để đáp ứng các nhu cầu này, c) đánh giá hiệu lực của các hành động được thực hiện,
d) đảm bảo rằng người lao động nhận thức được mối liên quan và tầm quan trọng của các hoạt động của họ và họ đóng góp như thế nào đối với việc đạt được mục tiêu chất lượng, và
e) duy trì hồ sơ thích hợp về giáo dục, đào tạo, kỹ năng và kinh nghiệm chuyên môn (xem 4.2.4).
CHÚ THÍCH: Các văn bản pháp quy quốc gia hoặc khu vực có thể đòi hỏi tổ chức phải thiết lập những thủ tục dạng văn bản để xác định các nhu cầu đào tạo.
6.3. Cơ sở hạ tầng
Tổ chức phải xác định, cung cấp và duy trì cơ sở hạ tầng cần thiết để đạt được sự phù hợp đối với các yêu cầu về sản phẩm. Cơ sở hạ tầng bao gồm ví dụ như:
a) nhà cửa, không gian làm việc và các phương tiện kèm theo;
b) trang thiết bị (cả phần cứng và phần mềm) và
c) dịch vụ hỗ trợ (như vận chuyển hoặc trao đổi thông tin).
Tổ chức phải thiết lập các yêu cầu dạng văn bản đối với các hoạt động bảo dưỡng, kể cả tần suất thực hiện, khi những hoạt động đó hoặc khi việc không có chúng sẽ ảnh hưởng đến chất lượng sản phẩm.
Hồ sơ của hoạt động bảo dưỡng phải được lưu giữ (xem 4.2.4).
6.4. Môi trường làm việc
Tổ chức phải xác định và quản lý môi trường làm việc cần thiết để đạt được sự phù hợp đối với các yêu cầu của sản phẩm.
Các yêu cầu sau phải áp dụng.
a) Tổ chức phải thiết lập các yêu cầu dạng văn bản về sức khỏe, tình trạng sạch sẽ và quần áo của người lao động nếu sự tiếp xúc giữa họ với sản phẩm hoặc môi trường làm việc có thể có ảnh hưởng xấu tới chất lượng của sản phẩm (xem 7.5.1.2.1).
b) Nếu điều kiện của môi trường làm việc có thể có tác động xấu đối với chất lượng sản phẩm, tổ chức phải thiết lập các yêu cầu dạng văn bản đối với điều kiện của môi trường làm việc đó và các thủ tục dạng văn bản hoặc hướng dẫn công việc để theo dõi và kiểm soát điều kiện của môi trường làm việc (xem 7.5.1.2.1).
c) Tổ chức phải đảm bảo rằng mọi người lao động được yêu cầu làm việc tạm thời trong điều kiện môi trường đặc biệt của môi trường làm việc đều được đào tạo một cách phù hợp hoặc được giám sát bởi những người đã được đào tạo [xem 6.2.2 b)].
d) Nếu thích hợp, phải thiết lập và lập thành văn bản các thỏa thuận đặc biệt về kiểm soát sản phẩm bị nhiễm bẩn và có khả năng bị nhiễm bẩn nhằm ngăn ngừa sự nhiễm bẩn của sản phẩm khác, của môi trường làm việc hoặc người lao động (xem 7.5.3.1).
7. Tạo sản phẩm
7.1. Hoạch định việc tạo sản phẩm
Tổ chức phải lập kế hoạch và triển khai các quá trình cần thiết đối với việc tạo sản phẩm. Hoạch định việc tạo sản phẩm phải nhất quán với các yêu cầu của các quá trình khác của hệ thống quản lý chất lượng (xem 4.1).
Trong quá trình hoạch định việc tạo sản phẩm, khi thích hợp tổ chức phải xác định những điều sau đây:
a) các mục tiêu chất lượng và các yêu cầu đối với sản phẩm;
b) nhu cầu thiết lập các quá trình, tài liệu và việc cung cấp các nguồn lực cụ thể đối với sản phẩm;
c) các hoạt động kiểm tra xác nhận, xác nhận giá trị sử dụng, các hoạt động theo dõi, kiểm tra và thử nghiệm cụ thể cần thiết đối với sản phẩm và các chuẩn mực chấp nhận sản phẩm;
d) các hồ sơ cần thiết để cung cấp bằng chứng rằng các quá trình thực hiện và sản phẩm tạo thành đáp ứng các yêu cầu (xem 4.2.4).
Đầu ra của việc hoạch định phải được thể hiện phù hợp với phương pháp tác nghiệp của tổ chức.
Tổ chức phải thiết lập các yêu cầu dạng văn bản đối với việc quản lý các rủi ro trong toàn bộ quá trình tạo sản phẩm. Hồ sơ liên quan đến quản lý rủi ro phải được lưu giữ (xem 4.2.4).
CHÚ THÍCH 1: Tài liệu qui định các quá trình của hệ thống quản lý chất lượng (bao gồm cả các quá trình tạo sản phẩm) và các nguồn lực được sử dụng đối với một sản phẩm, dự án hay hợp đồng cụ thể có thể được coi như một kế hoạch chất lượng.
CHÚ THÍCH 2: Tổ chức phải áp dụng các yêu cầu nêu trong 7.3 để triển khai quá trình tạo sản phẩm.
CHÚ THÍCH 3: Xem ISO 14971 về hướng dẫn liên quan đến quản lý rủi ro.
7.2. Các quá trình liên quan đến khách hàng
7.2.1. Xác định các yêu cầu liên quan đến sản phẩm
Tổ chức phải xác định
a) yêu cầu do khách hàng đưa ra, gồm cả các yêu cầu về các hoạt động giao hàng và sau giao hàng;
b) yêu cầu không được khách hàng công bố nhưng cần thiết cho việc sử dụng cụ thể hoặc sử dụng dự kiến khi đã biết;
c) yêu cầu chế định và pháp luật liên quan đến sản phẩm, và
d) mọi yêu cầu bổ sung do tổ chức xác định.
7.2.2. Xem xét các yêu cầu liên quan đến sản phẩm
Tổ chức phải xem xét các yêu cầu liên quan đến sản phẩm. Việc xem xét này phải được tiến hành trước khi tổ chức cam kết cung cấp sản phẩm cho khách hàng (ví dụ như nộp đơn dự thầu, chấp nhận hợp đồng hay đơn đặt hàng, chấp nhận sự thay đổi trong hợp đồng hay đơn đặt hàng) và phải đảm bảo rằng
a) yêu cầu về sản phẩm được định rõ và được lập thành văn bản;
b) các yêu cầu trong hợp đồng hoặc đơn đặt hàng khác với những gì đã nêu trước đó phải được giải quyết; và
c) tổ chức có khả năng đáp ứng các yêu cầu đã định.
Phải duy trì hồ sơ các kết quả của việc xem xét và các hành động nảy sinh từ việc xem xét (xem 4.2.4). Khi khách hàng đưa ra các yêu cầu không bằng văn bản, các yêu cầu của khách hàng phải được tổ chức đó khẳng định trước khi chấp nhận.
Khi yêu cầu về sản phẩm thay đổi, tổ chức phải đảm bảo rằng các văn bản tương ứng được sửa đổi và các cá nhân liên quan nhận thức được các yêu cầu thay đổi đó.
CHÚ THÍCH: Trong một số tình huống, ví dụ như trong bán hàng qua internet, với mỗi lần đặt hàng, việc xem xét một cách chính thức là không thực tế. Thay vào đó, việc xem xét có thể được thực hiện đối với các thông tin thích hợp về sản phẩm như danh mục chào hàng hay tài liệu quảng cáo.
7.2.3. Trao đổi thông tin với khách hàng
Tổ chức phải xác định và sắp xếp có hiệu quả việc trao đổi thông tin với khách hàng có liên quan tới a) thông tin về sản phẩm;
b) xử lý các yêu cầu, hợp đồng hoặc đơn đặt hàng, kể cả các sửa đổi;
c) phản hồi của khách hàng, kể cả các khiếu nại;
d) các thông báo tư vấn (xem 8.5.1).
7.3. Thiết kế và phát triển
7.3.1. Hoạch định thiết kế và phát triển
Tổ chức phải thiết lập các thủ tục dạng văn bản đối với thiết kế và phát triển.
Tổ chức phải lập kế hoạch và kiểm soát việc thiết kế và phát triển sản phẩm.
Trong quá trình hoạch định thiết kế và phát triển tổ chức phải xác định
a) các giai đoạn của thiết kế và phát triển,
b) các hoạt động xem xét, kiểm tra xác nhận, xác nhận giá trị sử dụng và chuyển giao thiết kế (xem CHÚ THÍCH) thích hợp cho mỗi giai đoạn thiết kế và phát triển, và
c) trách nhiệm và quyền hạn đối với các hoạt động thiết kế và phát triển.
Tổ chức phải quản lý sự tương giao giữa các nhóm khác nhau tham dự vào việc thiết kế và phát triển nhằm đảm bảo sự trao đổi thông tin có hiệu quả và phân công trách nhiệm rõ ràng.
Hoạch định đầu ra phải được lập thành văn bản và được cập nhật theo tiến triển của hoạt động thiết kế và phát triển (xem 4.2.3).
CHÚ THÍCH: Hoạt động chuyển giao thiết kế trong quá trình thiết kế và phát triển đảm bảo rằng đầu ra của thiết kế và phát triển được kiểm tra xác nhận là thích hợp với chế tạo sau khi trở thành quy định sản xuất cuối cùng.
7.3.2. Đầu vào của thiết kế và phát triển
Đầu vào liên quan đến các yêu cầu đối với sản phẩm phải được xác định và duy trì hồ sơ (xem 4.2.4).
Đầu vào phải bao gồm
a) yêu cầu về chức năng, hoạt động và an toàn theo công dụng,
b) yêu cầu chế định và luật pháp thích hợp,
c) thông tin có thể áp dụng nhận được từ các thiết kế tương tự trước đó, và
d) các yêu cầu khác cốt yếu cho thiết kế và phát triển,
e) đầu ra của quản lý rủi ro (xem 7.1).
Đầu vào này phải được xem xét về sự phù hợp và phải được chấp thuận.
Những đầu vào này phải được xem xét về sự thích đáng. Những yêu cầu này phải đầy đủ, không mơ hồ và không mâu thuẫn với nhau.
7.3.3. Đầu ra của thiết kế và phát triển
Đầu ra của thiết kế và phát triển phải ở dạng sao cho có thể kiểm tra xác nhận theo đầu vào của thiết kế và phát triển và phải được phê duyệt trước khi ban hành.
Đầu ra của thiết kế và phát triển phải:
a) đáp ứng các yêu cầu đầu vào của thiết kế và phát triển;
b) cung cấp các thông tin thích hợp cho việc mua hàng, sản xuất và cung cấp dịch vụ;
c) bao gồm hoặc viện dẫn tới các chuẩn mực chấp nhận của sản phẩm;
d) xác định các đặc tính cốt yếu cho an toàn và sử dụng đúng của sản phẩm.
Các hồ sơ về đầu ra của thiết kế và phát triển phải được lưu giữ (xem 4.2.4).
CHÚ THÍCH: Các hồ sơ về đầu ra của thiết kế và phát triển có thể bao gồm quy định kỹ thuật, quy trình chế tạo, bản vẽ kỹ thuật và nhật ký kỹ thuật hoặc nghiên cứu.
7.3.4. Xem xét thiết kế và phát triển
Tại những giai đoạn thích hợp, việc xem xét thiết kế và phát triển một cách có hệ thống phải được thực hiện theo hoạch định để
a) đánh giá khả năng đáp ứng các yêu cầu của các kết quả thiết kế và phát triển, và
b) nhận biết mọi vấn đề trục trặc và đề xuất các hành động cần thiết.
Những người tham dự vào việc xem xét phải bao gồm đại diện của tất cả các bộ phận chức năng liên quan tới các giai đoạn thiết kế và phát triển đang được xem xét cũng như các chuyên gia khác.(xem 5.5.1 và 6.2.1).
Phải duy trì hồ sơ về các kết quả xem xét và mọi hành động cần thiết (xem 4.2.4).
7.3.5. Kiểm tra xác nhận thiết kế và phát triển
Việc kiểm tra xác nhận phải được thực hiện theo các bố trí đã hoạch định (xem 7.3.1) để đảm bảo rằng đầu ra thiết kế và phát triển đáp ứng các yêu cầu đầu vào của thiết kế và phát triển.
Phải duy trì hồ sơ các kết quả kiểm tra xác nhận và duy trì mọi hoạt động cần thiết (xem 4.2.4).
7.3.6. Xác nhận giá trị sử dụng của thiết kế và phát triển
Xác nhận giá trị sử dụng của thiết kế và phát triển phải được tiến hành theo các bố trí đã hoạch định (xem 7.3.1) để đảm bảo rằng sản phẩm tạo ra có khả năng đáp ứng các yêu cầu sử dụng dự kiến hay các ứng dụng qui định khi đã biết. Phải tiến hành xác nhận giá trị sử dụng trước khi chuyển giao hay sử dụng sản phẩm (xem Chú thích 1).
Phải duy trì hồ sơ các kết quả của việc xác nhận giá trị sử dụng và mọi hành động cần thiết (xem 4.2.4).
Tổ chức phải thực hiện các đánh giá về khả năng sử dụng và/hoặc đánh giá về tính năng của dụng cụ y tế như là một hoạt động thành phần của việc xác nhận giá trị sử dụng của thiết kế và phát triển, theo yêu cầu của các văn bản pháp quy quốc gia hoặc khu vực (xem Chú thích 2).
CHÚ THÍCH 1: Nếu dụng cụ y tế chỉ có thể được xác nhận giá trị sử dụng sau khi lắp ráp và lắp đặt tại nơi sử dụng, việc chuyển giao chưa được coi là hoàn thành cho tới khi sản phẩm này đã chính thức được giao cho khách hàng.
CHÚ THÍCH 2: Việc cung cấp dụng cụ y tế cho mục đích đánh giá khả năng sử dụng và/hoặc đánh giá tính năng không được coi là hoạt động chuyển giao sản phẩm.
7.3.7. Kiểm soát thay đổi thiết kế và phát triển
Những thay đổi của thiết kế và phát triển phải được nhận biết và duy trì hồ sơ. Những thay đổi này phải được xem xét, kiểm tra xác nhận và xác nhận giá trị sử dụng một cách thích hợp và được phê duyệt trước khi thực hiện. Việc xem xét các thay đổi thiết kế và phát triển phải bao gồm việc đánh giá tác động của sự thay đổi lên các bộ phận cấu thành và sản phẩm đã được chuyển giao.
Phải duy trì hồ sơ các kết quả của việc xem xét các thay đổi và hành động cần thiết (xem 4.2.4).
7.4. Mua hàng
7.4.1. Quá trình mua hàng
Tổ chức phải thiết lập các thủ tục dạng văn bản để đảm bảo rằng sản phẩm mua vào phù hợp với các yêu cầu mua sản phẩm đã qui định.
Cách thức và mức độ kiểm soát áp dụng cho người cung ứng và sản phẩm mua vào phụ thuộc vào sự tác động của sản phẩm mua vào đối với việc tạo ra sản phẩm tiếp theo hay thành phẩm.
Tổ chức phải đánh giá và lựa chọn người cung ứng dựa trên khả năng cung cấp sản phẩm phù hợp với các yêu cầu của tổ chức. Phải xác định các chuẩn mực lựa chọn, đánh giá và đánh giá lại. Phải duy trì hồ sơ các kết quả của việc đánh giá và mọi hành động cần thiết nảy sinh từ việc đánh giá (xem 4.2.4).
7.4.2. Thông tin mua hàng
Thông tin mua hàng phải miêu tả sản phẩm được mua, nếu thích hợp có thể bao gồm:
a) yêu cầu về phê duyệt sản phẩm, các thủ tục, quá trình, và thiết bị;
b) yêu cầu về trình độ con người;
c) yêu cầu về hệ thống quản lý chất lượng.
Tổ chức phải đảm bảo sự thỏa đáng của các yêu cầu mua hàng đã qui định trước khi thông báo cho người cung ứng.
Theo mức độ yêu cầu về xác định nguồn gốc nêu trong 7.5.3.2, tổ chức phải duy trì các thông tin mua hàng liên quan, nghĩa là các tài liệu (xem 4.2.3) và hồ sơ (xem 4.2.4).
7.4.3. Kiểm tra xác nhận sản phẩm mua vào
Tổ chức phải lập và thực hiện các hoạt động kiểm tra hoặc các hoạt động khác cần thiết để đảm bảo rằng sản phẩm mua vào đáp ứng các yêu cầu mua hàng đã qui định.
Khi tổ chức hoặc khách hàng có ý định thực hiện các hoạt động kiểm tra xác nhận tại cơ sở của người cung ứng, tổ chức phải công bố việc sắp xếp kiểm tra xác nhận dự kiến và phương pháp thông qua sản phẩm trong các thông tin mua hàng.
Các hồ sơ của kiểm tra xác nhận phải được lưu giữ (xem 4.2.4).
7.5. Sản xuất và cung cấp dịch vụ
7.5.1. Kiểm soát sản xuất và cung cấp dịch vụ
7.5.1.1. Yêu cầu chung
Tổ chức phải lập kế hoạch, tiến hành sản xuất và cung cấp dịch vụ trong điều kiện được kiểm soát. Khi có thể, các điều kiện được kiểm soát phải bao gồm:
a) sự sẵn có các thông tin mô tả các đặc tính của sản phẩm,
b) sự sẵn có các hướng dẫn công việc khi cần,
c) việc sử dụng các thiết bị thích hợp,
d) sự sẵn có và việc sử dụng các phương tiện theo dõi và đo lường,
e) thực hiện việc đo lường và theo dõi,
f) thực hiện các hoạt động thông qua, giao hàng và các hoạt động sau giao hàng;
g) thực hiện các thao tác đã xác định về ghi nhãn và bao gói.
Tổ chức phải thiết lập và duy trì hồ sơ (xem 4.2.4) đối với từng mẻ dụng cụ y tế để xác định nguồn gốc ở mức độ quy định ở 7.5.3 và xác định lượng sản phẩm chế tạo và lượng sản phẩm chấp thuận phân phối. Hồ sơ về mẻ sản phẩm phải được kiểm tra xác nhận và phê duyệt.
CHÚ THÍCH: Một mẻ có thể là một dụng cụ y tế đơn lẻ.
7.5.1.2. Kiểm soát sản xuất và cung cấp dịch vụ - Yêu cầu riêng
7.5.1.2.1. Độ sạch của sản phẩm và kiểm soát sự nhiễm bẩn
Tổ chức phải thiết lập các yêu cầu dạng văn bản đối với độ sạch của sản phẩm nếu
a) sản phẩm được tổ chức làm sạch trước khi khử trùng và/hoặc sử dụng, hoặc
b) sản phẩm được cung ứng ở trạng thái chưa khử trùng và sẽ được đưa vào quá trình làm sạch trước khi khử trùng và/hoặc sử dụng, hoặc
c) sản phẩm được cung ứng để sử dụng ở trạng thái chưa khử trùng và độ sạch của sản phẩm là rất quan trọng trong sử dụng, hoặc
d) các tác nhân của quá trình được tách khỏi sản phẩm trong quá trình chế tạo.
Nếu sản phẩm được làm sạch theo a) hoặc b) nêu ở trên, các yêu cầu ở 6.4 a) và 6.4 b) không áp dụng trước cho quá trình làm sạch.
7.5.1.2.2. Hoạt động lắp đặt
Nếu phù hợp, tổ chức phải thiết lập các yêu cầu dạng văn bản trong đó có những chuẩn cứ chấp nhận đối với việc lắp đặt và kiểm tra xác nhận việc lắp đặt dụng cụ y tế.
Nếu yêu cầu đã được khách hàng đồng ý cho phép tổ chức khác hoặc đơn vị được tổ chức đó ủy quyền, thực hiện việc lắp đặt, tổ chức phải cung cấp các yêu cầu dạng văn bản đối với lắp đặt và kiểm tra xác nhận.
Hồ sơ về lắp đặt và kiểm tra xác nhận do tổ chức hoặc đơn vị được tổ chức ủy quyền thực hiện phải được lưu giữ (xem 4.2.4).
7.5.1.2.3. Hoạt động dịch vụ
Nếu việc cung cấp dịch vụ là yêu cầu quy định thì tổ chức phải thiết lập các thủ tục dạng văn bản, hướng dẫn công việc, tài liệu tham chiếu và quy trình đo chuẩn cần thiết để thực hiện hoạt động dịch vụ và kiểm tra xác nhận sự đáp ứng yêu cầu quy định của hoạt động dịch vụ.
Hồ sơ về hoạt động dịch vụ do tổ chức thực hiện phải được lưu giữ (xem 4,2,4).
CHÚ THÍCH: Ví dụ, dịch vụ có thể bao gồm sửa chữa và bảo dưỡng.
7.5.1.3. Yêu cầu cụ thể đối với dụng cụ y tế vô trùng
Tổ chức phải duy trì hồ sơ về các tham số của quá trình đối với quá trình khử trùng đã được sử dụng cho từng mẻ khử trùng (xem 4.2.4). Hồ sơ về khử trùng phải có khả năng xác định nguồn gốc đối với từng mẻ sản xuất dụng cụ y tế (xem 7.5.1.1).
7.5.2. Xác nhận giá trị sử dụng của các quá trình sản xuất và cung cấp dịch vụ
7.5.2.1. Yêu cầu chung
Tổ chức phải xác nhận giá trị sử dụng đối với của mọi quá trình sản xuất và cung cấp dịch vụ có kết quả đầu ra không thể kiểm tra xác nhận bằng cách theo dõi hoặc đo lường sau đó. Điều này bao gồm mọi quá trình mà sự sai sót chỉ có thể trở nên rõ ràng sau khi sản phẩm được sử dụng hoặc dịch vụ được chuyển giao.
Việc xác nhận giá trị sử dụng phải chứng tỏ khả năng của các quá trình để đạt được kết quả đã hoạch định.
Đối với các quá trình đó, khi có thể, tổ chức phải sắp xếp những điều sau:
a) các chuẩn mực đã định để xem xét và phê duyệt các quá trình;
b) phê duyệt thiết bị và trình độ con người;
c) sử dụng các phương pháp và thủ tục cụ thể;
d) các yêu cầu về hồ sơ (xem 4.2.4);
e) tái xác nhận giá trị sử dụng.
Tổ chức phải thiết lập các thủ tục dạng văn bản đối với việc xác nhận giá trị sử dụng của ứng dụng phần mềm máy tính (và những thay đổi đối với phần mềm đó và/hoặc ứng dụng của nó) đối với việc sản xuất và cung cấp dịch vụ mà có ảnh hưởng tới khả năng phù hợp với yêu cầu quy định của sản phẩm. ứng dụng phần mềm đó phải được xác nhận giá trị sử dụng trước khi sử dụng lần đầu.
Hồ sơ về xác nhận giá trị sử dụng phải được lưu giữ (xem 4.2.4).
7.5.2.2. Yêu cầu cụ thể đối với dụng cụ y tế vô trùng
Tổ chức phải thiết lập các thủ tục dạng văn bản đối với việc xác nhận giá trị sử dụng của các quá trình khử trùng. Quá trình khử trùng phải được xác nhận giá trị sử dụng trước khi sử dụng lần đầu.
Hồ sơ về xác nhận giá trị sử dụng của mỗi quá trình khử trùng phải được lưu giữ (xem 4.2.4).
7.5.3. Nhận biết và xác định nguồn gốc
7.5.3.1. Nhận biết
Tổ chức phải nhận biết sản phẩm bằng các biện pháp thích hợp trong suốt quá trình tạo sản phẩm và phải thiết lập thủ tục dạng văn bản cho việc nhận biết sản phẩm.
Tổ chức phải thiết lập các thủ tục dạng văn bản để đảm bảo rằng dụng cụ y tế được hoàn trả lại tổ chức sẽ được nhận biết và phân biệt được với sản phẩm phù hợp [xem 6.4 d)].
7.5.3.2. Xác định nguồn gốc
7.5.3.2.1. Quy định chung
Tổ chức phải thiết lập các thủ tục dạng văn bản đối với việc xác định nguồn gốc. Những thủ tục đó phải xác định mức độ xác định nguồn gốc của sản phẩm và các hồ sơ yêu cầu (xem 4.2.4, 8.3 và 8.5).
Tổ chức phải kiểm soát và lưu hồ sơ việc nhận biết duy nhất sản phẩm khi việc xác định nguồn gốc là một yêu cầu (xem 4.2.4).
CHÚ THÍCH: Quản lý cấu hình là phương pháp để duy trì việc nhận biết và xác định nguồn gốc.
7.5.3.2.2. Yêu cầu cụ thể đối với dụng cụ y tế cấy ghép hoạt tính và dụng cụ y tế cấy ghép
Để xác định các hồ sơ yêu cầu về xác định nguồn gốc, tổ chức phải có hồ sơ về tất cả các bộ phận cấu thành, vật liệu và điều kiện của môi trường làm việc nếu chúng là nguyên nhân làm cho dụng cụ y tế không thỏa mãn yêu cầu quy định.
Tổ chức phải yêu cầu các đại lý hoặc các nhà phân phối của mình duy trì hồ sơ về việc phân phối dụng cụ y tế để cho phép xác định nguồn gốc và những hồ sơ đó phải luôn sẵn có để phục vụ cho việc kiểm tra.
Hồ sơ về tên gọi và địa chỉ của bên nhận hàng phải được lưu giữ (xem 4.2.4).
7.5.3.3. Nhận biết trạng thái
Tổ chức phải nhận biết được trạng thái của sản phẩm tương ứng với các yêu cầu theo dõi và đo lường.
Việc nhận biết trạng thái của sản phẩm phải được duy trì trong suốt quá trình sản xuất, bảo quản, lắp đặt và cung cấp dịch vụ để đảm bảo rằng chỉ có sản phẩm đã qua kiểm tra và thử nghiệm (hoặc được nghiệm thu trong điều kiện nhượng bộ được phép) mới được gửi đi, sử dụng hoặc lắp đặt.
7.5.4. Tài sản của khách hàng
Tổ chức phải gìn giữ tài sản của khách hàng khi chúng thuộc sự kiểm soát của tổ chức hay được tổ chức sử dụng. Tổ chức phải nhận biết, kiểm tra xác nhận, bảo vệ tài sản do khách hàng cung cấp để sử dụng hoặc để hợp thành sản phẩm. Bất kỳ tài sản nào của khách hàng bị mất mát, hư hỏng hoặc được phát hiện không phù hợp cho việc sử dụng đều phải được thông báo cho khách hàng và các hồ sơ phải được duy trì (xem 4.2.4).
CHÚ THÍCH: Tài sản của khách hàng có thể bao gồm cả sở hữu trí tuệ.
7.5.5. Bảo toàn sản phẩm
Tổ chức phải thiết lập các thủ tục dạng văn bản hoặc hướng dẫn công việc dạng văn bản về bảo toàn sự phù hợp của sản phẩm trong suốt các quá trình nội bộ và giao hàng đến vị trí đã định.
Việc bảo toàn này phải bao gồm nhận biết, xếp dỡ (di chuyển), bao gói, lưu giữ, bảo quản. Việc bảo toàn cũng phải áp dụng với các bộ phận cấu thành của sản phẩm.
Tổ chức phải thiết lập các thủ tục dạng văn bản hoặc hướng dẫn công việc dạng văn bản về kiểm soát sản phẩm theo thời hạn sử dụng giới hạn hoặc điều kiện bảo quản đặc biệt yêu cầu. Điều kiện bảo quản đặc biệt đó phải được kiểm soát và lập thành hồ sơ (xem 4.2.4).
7.6. Kiểm soát phương tiện theo dõi và đo lường
Tổ chức phải xác định việc theo dõi và đo lường cần thực hiện và các phương tiện theo dõi và đo lường cần thiết để cung cấp bằng chứng về sự phù hợp của sản phẩm với các yêu cầu đã xác định (xem 7.2.1).
Tổ chức phải thiết lập các thủ tục dạng văn bản để đảm bảo rằng việc theo dõi và đo lường có thể tiến hành và được tiến hành một cách nhất quán với các yêu cầu theo dõi và đo lường.
Khi cần thiết để đảm bảo kết quả đúng, thiết bị đo lường phải:
a) được hiệu chuẩn hoặc kiểm tra xác nhận định kỳ, hoặc trước khi sử dụng, dựa trên các chuẩn đo lường có liên kết được với chuẩn đo lường quốc gia hay quốc tế; khi không có các chuẩn này thì căn cứ được sử dụng để hiệu chuẩn hoặc kiểm tra xác nhận phải được lưu hồ sơ;
b) được hiệu chỉnh hoặc hiệu chỉnh lại, khi cần thiết;
c) được nhận biết để giúp xác định trạng thái hiệu chuẩn;
d) được giữ gìn tránh bị hiệu chỉnh làm mất tính đúng đắn của các kết quả đo;
e) được bảo vệ để tránh hư hỏng hoặc suy giảm chất lượng trong khi di chuyển, bảo dưỡng và lưu giữ. Ngoài ra, tổ chức phải đánh giá và ghi nhận giá trị hiệu lực của các kết quả đo lường trước đó khi thiết bị được phát hiện không phù hợp với yêu cầu. Tổ chức phải tiến hành các hành động thích hợp đối với thiết bị đó và bất kỳ sản phẩm nào bị ảnh hưởng. Phải duy trì hồ sơ (xem 4.2.4) của kết quả hiệu chuẩn và kiểm tra xác nhận.
Khi sử dụng phần mềm máy tính để theo dõi và đo lường các yêu cầu đã qui định, phải khẳng định khả năng thỏa mãn việc áp dụng nhằm tới của chúng. Việc này phải được tiến hành trước lần sử dụng đầu tiên và được xác nhận lại khi cần thiết.
CHÚ THÍCH: Xem hướng dẫn trong ISO 10012 liên quan đến hệ thống quản lý đo lường.
8. Đo lường, phân tích và cải tiến
8.1. Khái quát
Tổ chức phải hoạch định và triển khai các quá trình theo dõi, đo lường, phân tích và cải tiến cần thiết để:
a) chứng tỏ sự phù hợp của sản phẩm;
b) đảm bảo sự phù hợp của hệ thống quản lý chất lượng;
c) duy trì tính hiệu lực của hệ thống quản lý chất lượng.
Điều này phải bao gồm việc xác định các phương pháp có thể áp dụng, kể cả các kỹ thuật thống kê, và mức độ sử dụng chúng.
CHÚ THÍCH: Các văn bản pháp quy quốc giá hoặc khu vực có thể yêu cầu những thủ tục dạng văn bản về áp dụng và kiểm soát việc ứng dụng các kỹ thuật thống kê.
8.2. Theo dõi và đo lường
8.2.1. Thông tin phản hồi
Tổ chức phải theo dõi các thông tin liên quan đến việc tổ chức có đáp ứng yêu cầu của khách hàng hay không, coi đó như một trong những thước đo mức độ thực hiện của hệ thống quản lý chất lượng.
Phải xác định các phương pháp để thu thập và sử dụng các thông tin này.
Tổ chức phải thiết lập thủ tục dạng văn bản đối với hệ thống phản hồi thông tin [xem 7.2.3 c)] để đưa ra cảnh báo sớm về các vấn đề chất lượng và đối với đầu vào của các quá trình của hành động khắc phục và phòng ngừa (xem 8.5.2 và 8.5.3).
Nếu các văn bản pháp quy quốc gia hoặc khu vực yêu cầu tổ chức phải tích luỹ kinh nghiệm của giai đoạn tiền sản xuất thì việc xem xét kinh nghiệm này phải là một phần của hệ thống phản hồi thông tin đó (xem 8.5.1).
8.2.2. Đánh giá nội bộ
Tổ chức phải tiến hành đánh giá nội bộ định kỳ theo kế hoạch để xác định xem hệ thống quản lý chất lượng:
a) có phù hợp với các bố trí sắp xếp được hoạch định (xem 7.1) đối với các yêu cầu của tiêu chuẩn này và với các yêu cầu của hệ thống chất lượng được tổ chức thiết lập, và
b) có được áp dụng một cách hiệu lực và được duy trì.
Tổ chức phải hoạch định chương trình đánh giá, có chú ý đến tình trạng và tầm quan trọng của các quá trình và các khu vực được đánh giá, cũng như kết quả của các cuộc đánh giá trước. Chuẩn mực, phạm vi, tần suất và phương pháp đánh giá phải được xác định. Việc lựa chọn các chuyên gia đánh giá và tiến hành đánh giá phải đảm bảo được tính khách quan và vô tư của quá trình đánh giá. Các chuyên gia đánh giá không được đánh giá công việc của mình.
Trách nhiệm và các yêu cầu về việc hoạch định và tiến hành các đánh giá, về việc báo cáo kết quả và duy trì hồ sơ (xem 4.2.4) phải được xác định trong một thủ tục dạng văn bản.
Lãnh đạo chịu trách nhiệm về khu vực được đánh giá phải đảm bảo tiến hành không chậm trễ các hành động để loại bỏ sự không phù hợp được phát hiện trong khi đánh giá và nguyên nhân của chúng.
Các hành động tiếp theo phải bao gồm việc kiểm tra xác nhận các hành động được tiến hành và báo cáo kết quả kiểm tra xác nhận (xem 8.5.2).
CHÚ THÍCH: Xem hướng dẫn trong ISO 19011 liên quan đến đánh giá chất lượng.
8.2.3. Theo dõi và đo lường các quá trình
Tổ chức phải áp dụng các phương pháp thích hợp cho việc theo dõi và, khi có thể, đo lường các quá trình của hệ thống quản lý chất lượng. Các phương pháp này phải chứng tỏ khả năng của các quá trình để đạt được các kết quả đã hoạch định. Khi không đạt được các kết quả theo hoạch định, phải tiến hành việc khắc phục và hành động khắc phục một cách thích hợp để đảm bảo sự phù hợp của sản phẩm.
8.2.4. Theo dõi và đo lường sản phẩm
8.2.4.1. Yêu cầu chung
Tổ chức phải theo dõi và đo lường các đặc tính của sản phẩm để kiểm tra xác nhận rằng các yêu cầu về sản phẩm được đáp ứng. Việc này phải được tiến hành tại những giai đoạn thích hợp của quá trình tạo sản phẩm theo các sắp xếp hoạch định (xem 7.1) và các thủ tục dạng văn bản (xem 7.5.1.1).
Bằng chứng của sự phù hợp với các chuẩn mực chấp nhận phải được duy trì. Hồ sơ phải chỉ ra người có quyền hạn trong việc thông qua sản phẩm (xem 4.2.4).
Chỉ được thông qua sản phẩm và chuyển giao dịch vụ khi đã hoàn thành thỏa đáng các hoạt động theo hoạch định (xem 7.1).
8.2.4.2. Yêu cầu cụ thể đối với dụng cụ y tế cấy ghép hoạt tính và dụng cụ y tế cấy ghép
Tổ chức phải lập hồ sơ (xem 4.2.4) nhân dạng của những người thực hiện các phép kiểm tra hoặc thử nghiệm.
8.3. Kiểm soát sản phẩm không phù hợp
Tổ chức phải đảm bảo rằng sản phẩm không phù hợp với các yêu cầu được nhận biết và kiểm soát để phòng ngừa việc sử dụng hoặc chuyển giao vô tình. Phải xác định trong một thủ tục dạng văn bản việc kiểm soát, các trách nhiệm và quyền hạn có liên quan đối với sản phẩm không phù hợp.
Tổ chức phải xử lý sản phẩm không phù hợp bằng một hoặc một số cách sau:
a) tiến hành loại bỏ sự không phù hợp được phát hiện;
b) cho phép sử dụng, thông qua hoặc chấp nhận có nhân nhượng;
c) tiến hành loại bỏ khỏi việc sử dụng hoặc áp dụng dự kiến ban đầu.
Tổ chức phải đảm bảo rằng sản phẩm không phù hợp chỉ được chấp nhận do nhân nhượng nếu đáp ứng yêu cầu chế định. Hồ sơ nhân dạng của những người cho phép nhân nhượng phải được lưu giữ (xem 4.2.4).
Phải duy trì hồ sơ (xem 4.2.4) về bản chất các sự không phù hợp và bất kỳ hành động tiếp theo nào được tiến hành, kể cả các nhân nhượng có được.
Khi sản phẩm không phù hợp được khắc phục, chúng phải được kiểm tra xác nhận lại để chứng tỏ sự phù hợp với các yêu cầu.
Khi sản phẩm không phù hợp được phát hiện sau khi chuyển giao hoặc đã bắt đầu sử dụng, tổ chức phải có các hành động thích hợp đối với các tác động hoặc hậu quả tiềm ẩn của sự không phù hợp.
Nếu cần phải làm lại sản phẩm (một lần hoặc nhiều lần), tổ chức phải lập thành văn bản quá trình làm lại đó dưới hình thức hướng dẫn công việc và tài liệu này phải trải qua cùng một thủ tục cho phép và phê duyệt như đối với hướng dẫn công việc ban đầu. Trước khi cho phép và phê duyệt hướng dẫn công việc đó, việc xác định mọi ảnh hưởng xấu của quá trình làm lại đối với sản phẩm phải được thực hiện và lập thành văn bản (xem 4.2.3 và 7.5.1).
8.4. Phân tích dữ liệu
Tổ chức phải thiết lập các thủ tục dạng văn bản để xác định, thu thập và phân tích các dữ liệu tương ứng để chứng tỏ sự thích hợp và tính hiệu lực của hệ thống quản lý chất lượng và đánh giá xem sự cải tiến hiệu lực của hệ thống chất lượng có thể tiến hành hay không.
Điều này bao gồm cả các dữ liệu được tạo ra do kết quả của việc theo dõi, đo lường và từ các nguồn thích hợp khác.
Việc phân tích dữ liệu phải cung cấp thông tin về:
a) thông tin phản hồi (xem 8.2.1);
b) sự phù hợp với các yêu cầu về sản phẩm (xem 7.2.1);
c) đặc tính và xu hướng của các quá trình và sản phẩm, kể cả các cơ hội cho hành động phòng ngừa;
d) người cung ứng.
Hồ sơ về kết quả phân tích dữ liệu phải được lưu giữ (xem 4.2.4).
8.5. Cải tiến
8.5.1. Khái quát
Tổ chức phải nhận biết và thực hiện mọi thay đổi cần thiết để đảm bảo và duy trì sự phù hợp liên tục và hiệu lực của hệ thống quản lý chất lượng thông qua việc sử dụng chính sách chất lượng, mục tiêu chất lượng, kết quả đánh giá, việc phân tích dữ liệu, hành động khắc phục và phòng ngừa và sự xem xét của lãnh đạo.
Tổ chức phải thiết lập các thủ tục dạng văn bản đối với việc ban hành và áp dụng các thông báo tư vấn. Các thủ tục đó phải có khả năng áp dụng ở mọi lúc.
Hồ sơ của tất cả các cuộc điều tra về khiếu nại của khách hàng đều phải được lưu giữ (xem 4.2.4). Nếu cuộc điều tra xác định rằng hoạt động bên ngoài tổ chức dẫn tới khiếu nại của khách hàng thì thông tin liên quan phải được trao đổi giữa các tổ chức liên quan (xem 4.1).
Nếu sau bất kỳ khiếu nại nào của khách hàng mà không có hành động khắc phục và/hoặc phòng ngừa thì nguyên nhân của việc không thực hiện hành động này phải được xác nhận (xem 5.5.1) và lập thành hồ sơ (4.2.4).
Nếu các văn bản pháp quy quốc gia hoặc khu vực yêu cầu phải thông báo về những sự việc có ảnh hưởng xấu đáp ứng chuẩn cứ thông báo quy định, tổ chức phải thiết lập các thủ tục dạng văn bản đối với việc thông báo đến các cơ quan quản lý có thẩm quyền.
8.5.2. Hành động khắc phục
Tổ chức phải thực hiện hành động nhằm loại bỏ nguyên nhân của sự không phù hợp để ngăn ngừa sự tái diễn. Hành động khắc phục phải tương ứng với tác động của sự không phù hợp gặp phải.
Phải lập một thủ tục dạng văn bản để xác định các yêu cầu về:
a) việc xem xét sự không phù hợp (kể cả các khiếu nại của khách hàng);
b) việc xác định nguyên nhân của sự không phù hợp;
c) việc đánh giá cần có các hành động để đảm bảo rằng sự không phù hợp không tái diễn;
d) việc xác định và thực hiện các hành động cần thiết, bao gồm, nếu phù hợp, việc cập nhật hệ thống tài liệu;
e) việc lưu hồ sơ các kết quả của mọi cuộc điều tra và hành động được thực hiện (xem 4.2.4);
f) việc xem xét các hành động khắc phục đã thực hiện và hiệu lực của hành động này.
8.5.3. Hành động phòng ngừa
Tổ chức phải xác định các hành động nhằm loại bỏ nguyên nhân của sự không phù hợp tiềm ẩn để ngăn chặn sự xuất hiện của chúng. Các hành động phòng ngừa được tiến hành phải tương ứng với tác động của các vấn đề tiềm ẩn.
Phải lập một thủ tục dạng văn bản để xác định các yêu cầu đối với:
a) việc xác định sự không phù hợp tiềm ẩn và các nguyên nhân của chúng;
b) việc đánh giá nhu cầu thực hiện các hành động để phòng ngừa việc xuất hiện sự không phù hợp;
c) việc xác định và thực hiện các hành động cần thiết;
d) việc lưu hồ sơ các kết quả của mọi cuộc điều tra và hành động được thực hiện (xem 4.2.4);
e) việc xem xét các hành động phòng ngừa được thực hiện và hiệu lực của hành động này.
PHỤ LỤC A
(tham khảo)
TƯƠNG ỨNG GIỮA TCVN ISO 13485 : 2004 VÀ ISO 13485 : 1996
Bảng A.1 - Tương ứng giữa ISO 13485 : 1996 và TCVN ISO 13485 : 2004
ISO 13485 : 1996 | TCVN ISO 13485 : 2004 |
1. Phạm vi áp dụng | 1 |
2. Tài liệu viện dẫn | 2 |
3. Định nghĩa | 3 |
4. Yêu cầu đối với hệ thống quản lý chất lượng [chỉ có tiêu đề] |
|
4.1. Trách nhiệm của lãnh đạo [chỉ có tiêu đề] 4.1.1. Chính sách chất lượng 4.1.2. Tổ chức [chỉ có tiêu đề] 4.1.2.1. Trách nhiệm và quyền hạn 4.1.2.2. Nguồn lực 4.1.2.3. Đại diện của lãnh đạo 4.1.3. Xem xét của lãnh đạo |
5.1 + 5.3 + 5.4.1
5.5.1 6.1 + 6.2.1 5.5.2 5.6.1 + 8.5.1 |
4.2. Hệ thống chất lượng [chỉ có tiêu đề] 4.2.1. Khái quát 4.2.2. Các thủ tục của hệ thống chất lượng 4.2.3. Hoạch định chất lượng |
4.1 + 4.2.2 4.2.1 5.4.2 + 7.1 |
4.3. Xem xét hợp đồng [chỉ có tiêu đề] 4.3.1. Khái quát [chỉ có tiêu đề] 4.3.2. Xem xét 4.3.3. Bổ sung hợp đồng 4.3.4. Hồ sơ |
5.2 + 7.2.1 + 7.2.2 + 7.2.3 7.2.2 7.2.2 |
4.4. Kiểm soát thiết kế [chỉ có tiêu đề] 4.4.1. Khái quát [chỉ có tiêu đề] 4.4.2. Hoạch định thiết kế và phát triển 4.4.3. Giao diện tổ chức và kỹ thuật 4.4.4. Đầu vào của thiết kế 4.4.5. Đầu ra của thiết kế 4.4.6. Xem xét thiết kế 4.4.7. Kiểm tra xác nhận thiết kế |
7.3.1 7.3.1 7.2.1 + 7.3.2 7.3.3 7.3.4 7.3.5 |
Bảng A.1(tiếp theo)
ISO 13485 : 1996 | TCVN ISO 13485 : 2004 |
4.4.8. Xác nhận giá trị sử dụng của thiết kế 4.4.9. Thay đổi của thiết kế | 7.3.6 7.3.7 |
4.5. Kiểm soát tài liệu và dữ liệu [chỉ có tiêu đề] 4.5.1. Khái quát 4.5.2. Phê duyệt tài liệu và dữ liệu và ban hành 4.5.3. Thay đổi của tài liệu và dữ liệu |
4.2.3 4.2.3 4.2.3 |
4.6. Mua hàng [chỉ có tiêu đề] 4.6.1. Khái quát [chỉ có tiêu đề] 4.6.2. Đánh giá người thầu phụ 4.6.3. Dữ liệu mua hàng 4.6.4. Kiểm tra xác nhận sản phẩm mua vào |
7.4.1 7.4.2 7.4.3 |
4.7. Kiểm soát sản phẩm do khách hàng cung ứng | 7.5.4 |
4.8. Nhận biết và xác định nguồn gốc sản phẩm | 7.5.3 |
4.9. Kiểm soát quá trình | 6.3 + 6.4 + 7.5.1 + 7.5.2 |
4.10. Kiểm tra và thử nghiệm [chỉ có tiêu đề] 4.10.1. Khái quát 4.10.2. Kiểm tra và thử nghiệm khi tiếp nhận 4.10.3. Kiểm tra và thử nghiệm trong quá trình 4.10.4. Kiểm tra và thử nghiệm cuối cùng 4.10.5. Hồ sơ kiểm tra và thử nghiệm |
7.1 + 8.1 7.4.3 + 8.2.4 8.2.4 8.2.4 7.5.3 + 8.2.4 |
4.11. Kiểm soát thiết bị kiểm tra, đo lường và thử nghiệm [chỉ có tiêu đề] 4.11.1. Khái quát 4.11.2. Thủ tục kiểm soát | 7.6 7.6 |
4.12. Trạng thái kiểm tra và thử nghiệm | 7.5.3 |
4.13. Kiểm soát sản phẩm không phù hợp [chỉ có tiêu đề] 4.13.1 Khái quát 4.13.2 Xem xét và thanh lọc sản phẩm không phù hợp | 8.3 8.3 |
4.14. Hành động khắc phục và phòng ngừa [chỉ có tiêu đề] 4.14.1. Khái quát 4.14.2. Hành động khắc phục 4.14.3. Hành động phòng ngừa | 8.5.2 + 8.5.3 8.5.2 8.5.3 |
Bảng A.1 (kết thúc)
ISO 13485 : 1996 | TCVN ISO 13485 : 2004 |
4.15. Vận chuyển, bảo quản, bao gói, bảo toàn và giao hàng [chỉ có tiêu đề] 4.15.1. Khái quát 4.15.2. Vận chuyển 4.15.3. Bảo quản 4.15.4. Bao gói 4.15.5. Bảo toàn 4.15.6. Giao hàng | 6.4 7.5.5 7.5.5 7.5.5 7.5.5 7.5.1 |
4.16. Kiểm soát hồ sơ chất lượng | 4.2.4 |
4.17. Đánh giá chất lượng nội bộ | 8.2.2 + 8.2.3 |
4.18. Đào tạo | 6.2.2 |
4.19. Dịch vụ bảo dưỡng | 7.5.1 |
4.20. Kỹ thuật thống kê [chỉ có tiêu đề] 4.20.1. Nhận biết nhu cầu 4.20.2. Các thủ tục |
8.1 + 8.2.3 + 8.2.4 + 8.4 8.1 + 8.2.3 + 8.2.4 + 8.4 |
Bảng A.2 - Tương ứng giữa TCVN ISO 13485 : 2004 và ISO 13485 : 1996
TCVN ISO 13485 : 2004 | ISO 13485 : 1996 |
1. Phạm vi áp dụng | 1 |
1.1. Khái quát |
|
1.2. Áp dụng |
|
2. Tài liệu viện dẫn | 2 |
3. Thuật ngữ và định nghĩa | 3 |
4. Hệ thống quản lý chất lượng [chỉ có tiêu đề] |
|
4.1. Yêu cầu chung | 4.2.1 |
4.2. Yêu cầu đối với hệ thống tài liệu [chỉ có tiêu đề] 4.2.1. Khái quát 4.2.2. Sổ tay chất lượng 4.2.3. Kiểm soát tài liệu 4.2.4. Kiểm soát hồ sơ |
4.2.2 4.2.1 4.5.1 + 4.5.2 + 4.5.3 4.16 |
5. Trách nhiệm của lãnh đạo [chỉ có tiêu đề] |
|
5.1. Cam kết của lãnh đạo | 4.1.1 |
5.2. Hướng vào khách hàng | 4.3.2 |
5.3. Chính sách chất lượng | 4.1.1 |
5.4. Hoạch định [chỉ có tiêu đề] |
|
5.4.1. Mục tiêu chất lượng 5.4.2. Hoạch định hệ thống quản lý chất lượng | 4.1.1 4.2.3 |
5.5. Trách nhiệm, quyền hạn và trao đổi thông tin [chỉ có tiêu đề] 5.5.1. Trách nhiệm và quyền hạn 5.5.2. Đại diện của lãnh đạo 5.5.3. Trao đổi thông tin nội bộ | 4.1.2.1 4.1.2.3 |
5.6. Xem xét của lãnh đạo [chỉ có tiêu đề] |
|
5.6.1. Khái quát 5.6.2. Đầu vào của việc xem xét 5.6.3. Đầu ra của việc xem xét | 4.1.3 |
6. Quản lý nguồn lực [chỉ có tiêu đề] |
|
6.1. Cung cấp nguồn lực | 4.1.2.2 |
6.2. Nguồn nhân lực [chỉ có tiêu đề] 6.2.1 Khái quát 6.2.2 Năng lực, nhận thức và đào tạo |
4.1.2.2 4.18 |
Bảng A.2(tiếp theo)
TCVN ISO 13485 : 2004 | ISO 13485 : 1996 |
6.3. Cơ sở hạ tầng | 4.9 |
6.4. Môi trường làm việc | 4.9 + 4.15.1 |
7. Tạo sản phẩm [chỉ có tiêu đề] |
|
7.1. Hoạch định việc tạo sản phẩm | 4.2.3 + 4.10.1 |
7.2. Các quá trình liên quan đến khách hàng [chỉ có tiêu đề] 7.2.1. Xác định các yêu cầu liên quan đến sản phẩm 7.2.2. Xem xét các yêu cầu liên quan đến sản phẩm 7.2.3. Trao đổi thông tin với khách hàng | 4.3.2 + 4.4.4 4.3.2 + 4.3.3 + 4.3.4 4.3.2 |
7.3. Thiết kế và phát triển [chỉ có tiêu đề] 7.3.1. Hoạch định thiết kế và phát triển 7.3.2. Đầu vào của thiết kế và phát triển 7.3.3. Đầu ra của thiết kế và phát triển 7.3.4. Xem xét thiết kế và phát triển 7.3.5. Kiểm tra xác nhận thiết kế và phát triển 7.3.6. Xác nhận giá trị sử dụng của thiết kế và phát triển 7.3.7. Kiểm soát thay đổi thiết kế và phát triển |
4.4.2 + 4.4.3 4.4.4 4.4.5 4.4.6 4.4.7 4.4.8 4.4.9 |
7.4. Mua hàng [chỉ có tiêu đề] |
|
7.4.1. Quá trình mua hàng 7.4.2. Thông tin mua hàng 7.4.3. Kiểm tra xác nhận sản phẩm mua vào | 4.6.2 4.6.3 4.6.4 + 4.10.2 |
7.5. Sản xuất và cung cấp dịch vụ [chỉ có tiêu đề] |
|
7.5.1. Kiểm soát sản xuất và cung cấp dịch vụ 7.5.2. Xác nhận giá trị sử dụng của các quá trình sản xuất và cung cấp dịch vụ 7.5.3. Nhận biết và xác định nguồn gốc 7.5.4. Tài sản của khách hàng 7.5.5. Bảo toàn sản phẩm | 4.9 + 4.15.6 + 4.19 4.9
4.7 4.15.2 + 4.15.3 + 4.15.4 + 4.15.5 |
7.6. Kiểm soát phương tiện theo dõi và đo lường | 4.10 + 4.20.1 + 4.20.2 |
Bảng A.2 (kết thúc)
TCVN ISO 13485 : 2004 | ISO 13485 : 1996 |
8. Đo lường, phân tích và cải tiến [chỉ có tiêu đề] |
|
8.1. Khái quát | 4.11.1 + 4.11.2 |
8.2. Theo dõi và đo lường 8.2.1. Thông tin phản hồi 8.2.2. Đánh giá nội bộ 8.2.3. Theo dõi và đo lường các quá trình 8.2.4. Theo dõi và đo lường sản phẩm |
4.17 4.17 + 4.20.1 + 4.20.2 4.10.2 + 4.10.3 + 4.10.4 + 4.10.5 + 4.20.1 + 4.20.2 |
8.3. Kiểm soát sản phẩm không phù hợp | 4.13.1 + 4.13.2 |
8.4. Phân tích dữ liệu | 4.20.1 + 4.20.2 |
8.5. Cải tiến [chỉ có tiêu đề] 8.5.1. Khái quát 8.5.2. Hành động khắc phục 8.5.3. Hành động phòng ngừa |
4.1.3 4.14.1 + 4.14.2 4.14.1 + 4.14.3 |
PHỤ LỤC B
(tham khảo)
GIẢI THÍCH VỀ NHỮNG KHÁC BIỆT GIỮA TCVN ISO 13485 : 2004 VÀ TCVN ISO 9001 : 2000
Phụ lục này lập bảng đối chiếu giữa những nội dung đồng nhất và khác biệt của tiêu chuẩn này và TCVN ISO 9001. Phụ lục này còn nêu ra luận chứng đối với những khác biệt giữa tiêu chuẩn này và TCVN ISO 9001.
a) Đối với những điều hoặc điều nhỏ của tiêu chuẩn này được trích dẫn trực tiếp và hoàn toàn không thay đổi so với TCVN ISO 9001 thì những điều nhỏ này được trình bày không thay đổi sẽ được chú thích trong tiêu chuẩn này bằng một đoạn văn đặt trong ngoặc vuông [].
b) Đối với những điều hoặc điều nhỏ của TCVN ISO 9001 được đưa với sự thay đổi vào tiêu chuẩn này bằng việc bổ sung thông tin hoặc bằng cách sửa đổi cho phù hợp với các quy định pháp lý liên quan đến dụng cụ y tế thì những điều hoặc điều nhỏ này của TCVN ISO 9001 được đưa nguyên văn vào cột bên trái của phụ lục này. Những điều hoặc điều nhỏ tương ứng của tiêu chuẩn này được đưa vào cột bên phải.
c) Đối với những điều hoặc điều nhỏ mà tiêu chuẩn này đã bỏ đi hoặc sửa đổi đoạn văn của TCVN ISO 9001 bằng cách bỏ đi hoặc thay đổi đáng kể yêu cầu có ý nghĩa quan trọng thì những điều hoặc điều nhỏ này của TCVN ISO 9001 được đưa nguyên văn vào cột bên trái của phụ lục này. Những điều hoặc điều nhỏ tương ứng của tiêu chuẩn này và lý do thay đổi được đưa vào cột bên phải.
d) Những lý do về sự khác biệt giữa tiêu chuẩn này và TCVN ISO 9001 được nêu ở cột bên phải. Khi không có "lý do về sự khác biệt" được nêu đối với mỗi điều hoặc điều nhỏ thì những khác biệt trong nội dung của hai tiêu chuẩn được xuất phát từ mục tiêu của TCVN ISO 13485 là phản ánh những quy định pháp lý hiện hành và thúc đẩy việc hài hòa các quy định pháp lý đối với dụng cụ y tế mới trên phạm vi toàn thế giới.
TCVN ISO 9001 : 2000 | TCVN ISO 13485 : 2004 | ||||||||
0. Giới thiệu 0.1. Khái quát Việc chấp nhận hệ thống quản lý chất lượng cần là một quyết định chiến lược của tổ chức. Việc thiết kế và áp dụng hệ thống quản lý chất lượng của một tổ chức phụ thuộc vào các nhu cầu khác nhau, các mục tiêu riêng biệt, các sản phẩm cung cấp, các quá trình được sử dụng, quy mô và cơ cấu của tổ chức. Tiêu chuẩn này không nhằm dẫn đến sự đồng nhất về cấu trúc của các hệ thống quản lý chất lượng hoặc của hệ thống tài liệu. Các yêu cầu của hệ thống quản lý chất lượng qui định trong tiêu chuẩn này bổ sung cho các yêu cầu đối với sản phẩm. Thông tin ở "CHÚ THÍCH" là để giải thích hoặc làm rõ các yêu cầu. Tiêu chuẩn này có thể được sử dụng cho nội bộ tổ chức và bên ngoài tổ chức, kể cả các tổ chức chứng nhận, để đánh giá khả năng đáp ứng các yêu cầu của khách hàng, yêu cầu chế định và yêu cầu riêng của tổ chức. Các nguyên tắc quản lý chất lượng nêu trong TCVN ISO 9004 đã được xem xét khi xây dựng tiêu chuẩn này. | 0. Giới thiệu 0.1. Khái quát Tiêu chuẩn này quy định các yêu cầu đối với hệ thống quản lý chất lượng có thể sử dụng bởi tổ chức thiết kế, phát triển, sản xuất, lắp đặt và bảo dưỡng dụng cụ y tế và bởi tổ chức thiết kế, phát triển và cung cấp dịch vụ liên quan. Tiêu chuẩn này còn áp dụng cho các tổ chức nội bộ và bên ngoài, kể cả các tổ chức chứng nhận, để đánh giá khả năng của các tổ chức đó đối với việc đáp ứng những yêu cầu chế định và yêu cầu của khách hàng. Thông tin ở "CHÚ THÍCH" là để giải thích hoặc làm rõ các yêu cầu. Lưu ý rằng các yêu cầu đối với hệ thống quản lý chất lượng quy định trong tiêu chuẩn này là để bổ sung cho những yêu cầu kỹ thuật đối với sản phẩm. Việc chấp nhận hệ thống quản lý chất lượng cần là một quyết định chiến lược của tổ chức. Việc thiết kế và áp dụng hệ thống quản lý chất lượng của một tổ chức phụ thuộc vào các nhu cầu khác nhau, các mục tiêu riêng biệt, các sản phẩm cung cấp, các quá trình được sử dụng, quy mô và cơ cấu của tổ chức. Tiêu chuẩn này không nhằm dẫn đến sự đồng nhất về cấu trúc của các hệ thống quản lý chất lượng hoặc của hệ thống tài liệu. Có rất nhiều chủng loại dụng cụ y tế và một số yêu cầu của tiêu chuẩn này chỉ áp dụng cho các nhóm dụng cụ y tế được nêu ra. Các nhóm này được xác định ở Điều 3. Lý do về sự khác biệt: Trừ những nội dung của đoạn 4 của 0.1 của TCVN ISO 13485, mọi thay đổi so với nội dung của 0.1 của TCVN ISO 9001 là chỉ nhằm áp dụng điều này cho dụng cụ y tế. | ||||||||
0.2. Cách tiếp cận theo quá trình Tiêu chuẩn này khuyến khích việc chấp nhận cách tiếp cận theo quá trình khi xây dựng, thực hiện và nâng cao hiệu lực của hệ thống quản lý chất lượng, nhằm thỏa mãn khách hàng qua việc đáp ứng yêu cầu của họ. Để hoạt động có hiệu quả, tổ chức phải xác định và quản lý nhiều hoạt động có liên quan mật thiết với nhau. Bất cứ hoạt động nào sử dụng nguồn lực và được quản lý nhằm chuyển hóa các đầu vào thành các đầu ra có thể được coi như một quá trình. Thông thường, đầu ra của một quá trình này sẽ trực tiếp là đầu vào của quá trình tiếp theo. Việc áp dụng hệ thống các quá trình trong một tổ chức, cùng với sự nhận biết các mối tương tác giữa các quá trình như vậy, và việc quản lý chúng, có thể được coi như "cách tiếp cận theo quá trình". Ưu thế của cách tiếp cận theo quá trình là kiểm soát công việc đang diễn ra, việc kiểm soát này bao trùm sự kết nối các quá trình đơn lẻ trong hệ thống các quá trình, cũng như bao trùm cả sự kết hợp và tương tác giữa các quá trình đó. Khi được sử dụng trong hệ thống quản lý chất lượng, cách tiếp cận trên nhấn mạnh tầm quan trọng của: a) việc hiểu và đáp ứng các yêu cầu; b) nhu cầu xem xét quá trình trong vấn đề giá trị gia tăng; c) có được kết quả về tính hiệu lực và hiệu quả của quá trình; d) cải tiến liên tục quá trình trên cơ sở đo lường đối tượng. Mô hình hệ thống quản lý chất lượng dựa trên quá trình nêu ở Hình 1 minh họa sự kết hợp các quá trình được trình bày trong điều 4 đến điều 8. Mô hình cho thấy khách hàng đóng vai trò quan trọng trong việc xác định các yêu cầu là đầu vào. Việc theo dõi sự thỏa mãn của khách hàng đòi hỏi có sự đánh giá các thông tin liên quan đến sự chấp nhận của khách hàng, chẳng hạn như liệu các yêu cầu của khách hàng có được đáp ứng không. Mô hình nêu ở hình 1 không phản ánh chi tiết các quá trình, nhưng bao quát tất cả các yêu cầu của tiêu chuẩn này. CHÚ THÍCH: Ngoài ra, phương pháp luận quen thuộc "Lập kế hoạch - Thực hiện - Kiểm tra - Hành động" (PDCA) có thể áp dụng cho mọi quá trình. Có thể mô tả tóm tắt PDCA như sau:
Hình 1 - Mô hình về hệ thống quản lý chất lượng dựa trên quá trình | 0.2. Cách tiếp cận theo quá trình Tiêu chuẩn này căn cứ vào cách tiếp cận theo quá trình đối với quản lý chất lượng. Hoạt động bất kỳ nhận đầu vào và chuyển hóa chúng thành đầu ra đều có thể được xem là quá trình. Để tổ chức hoạt động có hiệu lực, cần xác định và quản lý hàng loạt quá trình liên kết với nhau. Đầu ra của một quá trình thường trực tiếp là đầu vào của quá trình tiếp sau. Việc áp dụng hệ thống các quá trình trong một tổ chức, cùng với sự nhận biết các mối tương tác giữa các quá trình như vậy, và việc quản lý chúng, có thể được coi như "cách tiếp cận theo quá trình". Lý do về sự khác biệt: Phần lớn nội dung hướng dẫn nêu ở 0.2 của TCVN ISO 9001 đang được xem xét để đưa vào ISO/TR 14969, đó là tiêu chuẩn đưa ra hướng dẫn đối với việc áp dụng các yêu cầu của tiêu chuẩn này. Thông tin này được nêu trong 0.2 của TCVN ISO 9001 do tiêu chuẩn đó hiện không có tài liệu hướng dẫn kèm theo tương tự như ISO/TR 14969. Do ISO/TR 14969 đang được xây dựng nên nội dung hướng dẫn còn chưa được đưa vào 0.2 của TCVN ISO 13485. | ||||||||
| |||||||||
0.3. Mối quan hệ với TCVN ISO 9004 Ấn bản này của TCVN ISO 9001 và TCVN ISO 9004 được xây dựng như là một cặp thống nhất các tiêu chuẩn về hệ thống quản lý chất lượng. Hai tiêu chuẩn này được thiết kế để sử dụng đồng thời, nhưng cũng có thể được sử dụng một cách độc lập. Mặc dù hai tiêu chuẩn này có phạm vi khác nhau, nhưng chúng có cấu trúc tương tự để thuận tiện cho việc sử dụng như một cặp thống nhất. TCVN ISO 9001 qui định các yêu cầu đối với một hệ thống quản lý chất lượng, có thể được sử dụng trong nội bộ tổ chức sử dụng, cho việc chứng nhận hoặc cho các mục đích hợp đồng. Tiêu chuẩn tập trung vào hiệu quả của hệ thống quản lý chất lượng trong việc thỏa mãn yêu cầu khách hàng. So với TCVN ISO 9001, TCVN ISO 9004 đưa ra hướng dẫn với phạm vi rộng hơn về các mục tiêu của hệ thống quản lý chất lượng, đặc biệt đối với việc cải tiến liên tục hoạt động chung, hiệu quả của tổ chức cũng như hiệu lực của hệ thống này. TCVN ISO 9004 được khuyến nghị áp dụng như một tài liệu hướng dẫn đối với các tổ chức có lãnh đạo cao nhất mong muốn đạt được sự tiến triển dựa trên những yêu cầu của TCVN ISO 9001 nhằm cải tiến liên tục hoạt động của tổ chức. Tuy nhiên, điều này không nhằm vào mục đích chứng nhận hoặc hợp đồng. | 0.3. Mối quan hệ với các tiêu chuẩn khác 0.3.1. Mối quan hệ với TCVN ISO 9001:2000 Khi là tiêu chuẩn độc lập, tiêu chuẩn này căn cứ vào TCVN ISO 9001. Các điều hoặc quy định được trích dẫn trực tiếp và không thay đổi so với TCVN ISO 9001 được thể hiện bằng kiểu chữ đứng. Phụ lục B trình bày những điều và quy định thực sự không thay đổi này. Khi mà nội dung quy định của tiêu chuẩn này không tương đương với nội dung quy định của TCVN ISO 9001 thì toàn bộ câu hoặc đoạn có phần nội dung quy định đó được thể hiện bằng kiểu chữ in nghiêng (chữ in nghiêng màu xanh trong bản điện tử). Bản chất và nguyên nhân đối với những thay đổi trong nội dung quy định được nêu ở Phụ lục B. 0.3.2. Mối quan hệ với ISO/TR 14969 ISO/TR 14969 là báo cáo kỹ thuật cung cấp hướng dẫn cho việc áp dụng ISO 13485 (và TCVN ISO 13485). Lý do về sự khác biệt: Không hề có mối quan hệ đáng kể nào giữa tiêu chuẩn này và TCVN ISO 9004. Các mối quan hệ chính mang lại lợi ích từ sự giải thích trong phần giới thiệu này là những mối quan hệ giữa tiêu chuẩn này với TCVN ISO 9001 và ISO/TR 14969. | ||||||||
0.4. Sự tương thích với các hệ thống quản lý khác Tiêu chuẩn này được liên kết với TCVN ISO 14001:1996 nhằm tăng độ tương thích của hai tiêu chuẩn đối với lợi ích của cộng đồng người sử dụng. Tiêu chuẩn này không bao gồm các yêu cầu cụ thể cho các hệ thống quản lý khác, như các hệ thống quản lý môi trường, quản lý an toàn và sức khỏe nghề nghiệp, quản lý tài chính và rủi ro. Tuy nhiên, tiêu chuẩn này giúp tổ chức hòa hợp và hợp nhất hệ thống quản lý của mình với các yêu cầu của các hệ thống quản lý có liên quan. Điều này làm cho tổ chức có thể điều chỉnh hệ thống quản lý hiện hành của mình nhằm mục đích thiết lập một hệ thống quản lý chất lượng phù hợp với các yêu cầu của tiêu chuẩn này. | 0.4. Sự tương hợp với các hệ thống quản lý khác Tiêu chuẩn này được trình bày theo thể thức trình bày của TCVN ISO 9001 để tạo thuận tiện cho người sử dụng tiêu chuẩn trong cộng đồng những người sử dụng dụng cụ y tế. Tiêu chuẩn này không bao gồm các yêu cầu riêng đối với những hệ thống quản lý khác, chẳng hạn những yêu cầu cụ thể đối với hệ thống quản lý môi trường, hệ thống quản lý sức khỏe nghề nghiệp và an toàn hoặc hệ thống quản lý tài chính. Tuy nhiên, tiêu chuẩn này tạo điều kiện cho tổ chức tiệm cận hoặc kết hợp hệ thống quản lý chất lượng của mình với các yêu cầu liên quan về hệ thống quản lý. Điều này giúp cho tổ chức có khả năng chuyển đổi (các) hệ thống quản lý hiện hành của mình nhằm thiết lập hệ thống quản lý chất lượng phù hợp với yêu cầu của tiêu chuẩn này. Lý do về sự khác biệt: Đoạn đầu của 0.4 của TCVN ISO 13485 nhấn mạnh đến sự tiệm cận của TCVN ISO 13485 với TCVN ISO 9001. Quản lý rủi ro là một yêu cầu chính trong nhiều hoạt động và yêu cầu gắn kết với hệ thống quản lý chất lượng cho các tổ chức sản xuất và cung ứng dụng cụ y tế. | ||||||||
1. Phạm vi 1.1. Khái quát Tiêu chuẩn này quy định các yêu cầu đối với hệ thống quản lý chất lượng khi một tổ chức a) cần chứng tỏ khả năng cung cấp một cách ổn định sản phẩm đáp ứng các yêu cầu của khách hàng và các yêu cầu chế định thích hợp; b) nhằm để nâng cao sự thỏa mãn của khách hàng thông qua việc áp dụng có hiệu lực hệ thống, bao gồm cả các quá trình để cải tiến liên tục hệ thống và đảm bảo sự phù hợp với các yêu cầu của khách hàng và yêu cầu chế định được áp dụng CHÚ THÍCH - Trong tiêu chuẩn này, thuật ngữ "sản phẩm" chỉ áp dụng cho sản phẩm nhằm cho khách hàng hoặc khách hàng yêu cầu. | 1. Phạm vi áp dụng 1.1. Khái quát Tiêu chuẩn này quy định các yêu cầu đối với hệ thống quản lý chất lượng trong đó một tổ chức cần thể hiện khả năng cung cấp dụng cụ y tế và dịch vụ liên quan đáp ứng đúng yêu cầu của khách hàng và yêu cầu chế định áp dụng cho dụng cụ y tế và dịch vụ liên quan. Mục tiêu hàng đầu của tiêu chuẩn này là thúc đẩy áp dụng các yêu cầu chế định hài hòa về dụng cụ y tế đối với hệ thống quản lý chất lượng. Kết quả là tiêu chuẩn này bao gồm một số yêu cầu cụ thể đối với dụng cụ y tế và không bao gồm một số yêu cầu của TCVN ISO 9001 không phù hợp làm yêu cầu chế định. Do những ngoại lệ này, các tổ chức có hệ thống quản lý chất lượng phù hợp với tiêu chuẩn này không thể công bố sự phù hợp của chúng với TCVN ISO 9001 trừ khi chúng phù hợp với tất cả các yêu cầu của TCVN ISO 9001 (xem Phụ lục B). Lý do về sự khác biệt: Điều này sử dụng và giải thích các thuật ngữ thích hợp cho ngành dụng cụ y tế. Ngoài ra, các thuật ngữ "sự thỏa mãn của khách hàng" và "cải tiến liên tục" được loại bỏ vì chúng không phù hợp sử dụng trong tiêu chuẩn có mục tiêu là thúc đẩy việc hài hòa các văn bản pháp quy liên quan đến dụng cụ y tế về hệ thống quản lý chất lượng trên toàn thế giới. Đoạn 2 nhằm làm rõ mục đích của TCVN ISO 13485 là thúc đẩy việc hài hòa các yêu cầu chế định về hệ thống quản lý chất lượng trên toàn thế giới, chỉ ra rằng mục đích này đòi hỏi sự bổ sung thêm một vài yêu cầu không có trong TCVN ISO 9001 và bỏ đi một vài yêu cầu có trong TCVN ISO 9001 và làm rõ rằng việc gắn kết với TCVN ISO 13485 không hề đồng nghĩa với việc gắn kết với TCVN ISO 9001. Thuật ngữ "và các dịch vụ liên quan" được thêm vào hai lần và làm thay đổi thuật ngữ "dụng cụ y tế" là vì "dụng cụ y tế" không bao gồm "dịch vụ" trong định nghĩa. Ngược lại, trong khi đó ở TCVN ISO 9001, thuật ngữ "sản phẩm" bao gồm cả "dịch vụ" trong định nghĩa rồi. | ||||||||
1.2. Áp dụng Các yêu cầu trong tiêu chuẩn này mang tính tổng quát và nhằm để áp dụng cho mọi tổ chức không phân biệt vào loại hình, quy mô và sản phẩm cung cấp. Khi có yêu cầu nào đó của tiêu chuẩn này không thể áp dụng được do bản chất của tổ chức và sản phẩm của mình, có thể xem xét yêu cầu này như một ngoại lệ. Khi có ngoại lệ, việc được công bố phù hợp với tiêu chuẩn này không được chấp nhận trừ phi các ngoại lệ này được giới hạn trong phạm vi điều 7, và các ngoại lệ này không ảnh hưởng đến khả năng hay trách nhiệm của tổ chức trong việc cung cấp các sản phẩm đáp ứng các yêu cầu của khách hàng và các yêu cầu thích hợp. | 1.2. Áp dụng Tất cả các yêu cầu của tiêu chuẩn này đều là những yêu cầu áp dụng riêng cho những tổ chức cung cấp dụng cụ y tế, bất kể những tổ chức này thuộc loại hình hoặc có quy mô như thế nào. Nếu các yêu cầu chế định cho phép có các ngoại lệ về kiểm soát thiết kế và phát triển (xem 7.3) thì điều này có thể sử dụng để giải thích cho lý do đưa những ngoại lệ đó vào hệ thống quản lý chất lượng. Các văn bản pháp quy này có thể cung cấp những thỏa thuận khác sẽ được đề cập đến trong hệ thống quản lý chất lượng. Trách nhiệm của tổ chức là đảm bảo rằng những công bố về sự phù hợp với tiêu chuẩn này sẽ phản ánh sự ngoại lệ đối với kiểm soát thiết kế và phát triển [xem 4.2.2 a) và 7.3]. Nếu mọi yêu cầu trong Điều 7 của tiêu chuẩn này đều không áp dụng được do bản chất của dụng cụ y tế là đối tượng áp dụng của hệ thống quản lý chất lượng thì tổ chức không cần thiết phải đưa những yêu cầu đó vào hệ thống quản lý chất lượng của mình [xem 4.2.2 a)]. Các quá trình mà tiêu chuẩn này yêu cầu và áp dụng được cho dụng cụ y tế nhưng không được tổ chức thực hiện thì đó là trách nhiệm của tổ chức và chúng đều phải được giải thích trong hệ thống quản lý chất lượng của tổ chức [xem 4.1 a)]. Trong tiêu chuẩn này, các cụm từ "nếu thích hợp" và "ở nơi thích hợp" được sử dụng nhiều lần. Khi một yêu cầu được bổ nghĩa bởi các cụm từ này thì yêu cầu đó dường như là "thích hợp" trừ khi tổ chức có thể viết trong tài liệu cách giải thích khác. Một yêu cầu được xem là "thích hợp" nếu yêu cầu đó cần để cho: - sản phẩm đáp ứng các yêu cầu quy định; - tổ chức tiến hành hành động khắc phục. Lý do về sự khác biệt: Nội dung của điều này làm rõ rằng các yêu cầu của TCVN ISO 13485 là những yêu cầu đặc thù đối với ngành dụng cụ y tế. Ngoài ra, điều này còn nhấn mạnh mối quan hệ giữa việc cho ngoại lệ đối với thiết kế và phát triển mà yêu cầu này có thể có ảnh hưởng chế định ở một số nơi trên thế giới. Cuối cùng, điều này đưa ra sự phân biệt giữa các yêu cầu ở Điều 7, trong đó quy định một tổ chức có thể bằng sự biện minh chế định mà thực hiện ngoại lệ đối với hệ thống quản lý chất lượng của mình (giới hạn ở 7.3) ngay cả khi tổ chức đó có thể thực hiện những hoạt động liên quan đến các yêu cầu đó với những yêu cầu mà theo đó tổ chức có thể bằng sự biện minh mà không đưa vào hệ thống quản lý chất lượng của mình vì những yêu cầu đó liên quan đến các hoạt động mà tổ chức không thực hiện. | ||||||||
2. Tiêu chuẩn trích dẫn TCVN ISO 9000: 2000 (ISO 9000 : 2000) Hệ thống quản lý chất lượng - Cơ sở và từ vựng. | 2. Tiêu chuẩn viện dẫn TCVN ISO 9000 : 2000 (ISO 9000 : 2000) Hệ thống quản lý chất lượng - Cơ sở và từ vựng. Lý do về sự khác biệt: TCVN ISO 13485 được trình bày theo TCVN 1-2:2003 . | ||||||||
3. Thuật ngữ và định nghĩa Tiêu chuẩn này sử dụng các thuật ngữ và định nghĩa trong TCVN ISO 9000: 2000 . Các thuật ngữ sau, được sử dụng trong ấn bản này của TCVN ISO 9001 để mô tả chuỗi cung cấp, đã được thay đổi để phản ánh từ vựng được sử dụng hiện hành: người cung ứng → tổ chức → khách hàng Thuật ngữ "tổ chức" thay thế cho thuật ngữ "người cung ứng" được sử dụng trước đây trong TCVN ISO 9001: 1996 (TCVN ISO 9001: 1994) để chỉ đơn vị áp dụng tiêu chuẩn này. Thuật ngữ "người cung ứng" lúc này được sử dụng thay cho thuật ngữ "người thầu phụ". Trong tiêu chuẩn này, thuật ngữ "sản phẩm" cũng có nghĩa "dịch vụ". | 3. Thuật ngữ và định nghĩa Tiêu chuẩn này áp dụng các thuật ngữ và định nghĩa cho trong TCVN ISO 9000 và các thuật ngữ, định nghĩa dưới đây. Các thuật ngữ dưới đây, sử dụng trong tiêu chuẩn này để mô tả chuỗi cung ứng, đã được thay đổi để phản ánh từ vựng hiện hành: người cung ứng → tổ chức → khách hàng Thuật ngữ "tổ chức" thay thế cho thuật ngữ "người cung ứng" đã sử dụng trong ISO 13485:1996 và chỉ đơn vị áp dụng tiêu chuẩn này. Thuật ngữ "người cung ứng" hiện còn thay thế cho thuật ngữ "người thầu phụ". Trong tiêu chuẩn này, thuật ngữ "sản phẩm" cũng có nghĩa là "dịch vụ". Khi mà các yêu cầu được quy định để áp dụng cho "dụng cụ y tế" thì chúng cũng áp dụng đầy đủ cho những dịch vụ liên quan do tổ chức cung cấp. Các thuật ngữ dưới đây cần được coi là những thuật ngữ chung do các định nghĩa nêu trong những văn bản pháp quy quốc gia có thể khác biệt đôi chút và được ưu tiên hơn. Lý do về sự khác biệt: Nội dung của điều này thích hợp sử dụng trong ngành dụng cụ y tế và cũng bao gồm cả những quy định liên quan đến việc các văn bản pháp quy của nơi áp dụng có thể có định nghĩa thay thế cho định nghĩa có trong TCVN ISO 13485 hoặc được viện dẫn từ TCVN ISO 13485. Các định nghĩa 3.1 đến 3.8 là những định nghĩa đặc thù cho ngành dụng cụ y tế. | ||||||||
| |||||||||
4. Hệ thống quản lý chất lượng 4.1. Yêu cầu chung Tổ chức phải xây dựng, lập văn bản, thực hiện, duy trì hệ thống quản lý chất lượng và thường xuyên nâng cao hiệu lực của hệ thống theo các yêu cầu của tiêu chuẩn này. Tổ chức phải: a) nhận biết các quá trình cần thiết trong hệ thống quản lý chất lượng và áp dụng chúng trong toàn bộ tổ chức (xem 1.2), b) xác định trình tự và mối tương tác của các quá trình này, c) xác định các chuẩn mực và phương pháp cần thiết để đảm bảo việc tác nghiệp và kiểm soát các quá trình này có hiệu lực, d) đảm bảo sự sẵn có của các nguồn lực và thông tin cần thiết để hỗ trợ hoạt động tác nghiệp và theo dõi các quá trình này, e) đo lường, theo dõi và phân tích các quá trình này, và f) thực hiện các hành động cần thiết để đạt được kết quả dự định và cải tiến liên tục các quá trình này. Tổ chức phải quản lý các quá trình tuân thủ theo các yêu cầu của tiêu chuẩn này. Khi tổ chức chọn nguồn bên ngoài cho bất kỳ quá trình nào ảnh hưởng đến sự phù hợp của sản phẩm với các yêu cầu, tổ chức phải đảm bảo kiểm soát được những quá trình đó. Việc kiểm soát những quá trình do nguồn bên ngoài phải được nhận biết trong hệ thống quản lý chất lượng. CHÚ THÍCH: Các quá trình cần thiết đối với hệ thống quản lý chất lượng nêu ở trên cần bao gồm cả các quá trình về các hoạt động quản lý, cung cấp nguồn lực, tạo sản phẩm và đo lường. | 4. Hệ thống quản lý chất lượng 4.1. Yêu cầu chung Tổ chức phải xây dựng, lập văn bản, thực hiện, duy trì hệ thống quản lý chất lượng và duy trì hiệu lực của hệ thống theo các yêu cầu của tiêu chuẩn này. Tổ chức phải: a) nhận biết các quá trình cần thiết trong hệ thống quản lý chất lượng và áp dụng chúng trong toàn bộ tổ chức (xem 1.2); b) xác định trình tự và mối liên hệ của các quá trình; c) xác định chuẩn mực và phương pháp cần thiết để đảm bảo việc tác nghiệp và kiểm soát các quá trình có hiệu lực; d) đảm bảo sự sẵn có của các nguồn lực và thông tin cần thiết để hỗ trợ hoạt động tác nghiệp và theo dõi các quá trình này; e) đo lường, theo dõi và phân tích các quá trình; f) thực hiện các hành động cần thiết để đạt được kết quả dự định và duy trì hiệu lực của các quá trình này. Tổ chức phải quản lý các quá trình tuân thủ theo các yêu cầu của tiêu chuẩn. Khi tổ chức chọn nguồn bên ngoài cho bất kỳ quá trình nào ảnh hưởng đến sự phù hợp của sản phẩm với các yêu cầu, tổ chức phải đảm bảo kiểm soát được những quá trình đó. Việc kiểm soát những quá trình do nguồn bên ngoài phải được nhận biết trong hệ thống quản lý chất lượng (xem 8.5.1). CHÚ THÍCH: Các quá trình cần thiết đối với hệ thống quản lý chất lượng nêu trên cần bao gồm cả các quá trình hoạt động quản lý, cung cấp nguồn lực, tạo sản phẩm và đo lường. Lý do về sự khác biệt: Nội dung của điều này phù hợp với các mục tiêu của việc thực hiện quy định của các văn bản pháp quy hiện hành và của việc thúc đẩy sự hài hòa các văn bản pháp quy liên quan đến dụng cụ y tế trên phạm vi toàn thế giới. Các văn bản pháp quy hiện hành hướng vào tính hiệu lực của hệ thống quản lý chất lượng nhằm chỉ sản xuất ra những sản phẩm an toàn và có hiệu lực. | ||||||||
4.2. Yêu cầu về hệ thống tài liệu 4.2.1. Khái quát Các tài liệu của hệ thống quản lý chất lượng phải bao gồm a) các văn bản công bố về chính sách chất lượng và mục tiêu chất lượng, b) sổ tay chất lượng, c) các thủ tục dạng văn bản theo yêu cầu của tiêu chuẩn này, d) các tài liệu cần có của tổ chức để đảm bảo việc hoạch định, tác nghiệp và kiểm soát có hiệu lực các quá trình của tổ chức đó, và e) các hồ sơ theo yêu cầu của tiêu chuẩn này (xem 4.2.4). CHÚ THÍCH 1: Khi thuật ngữ "thủ tục dạng văn bản" xuất hiện trong tiêu chuẩn này, thì thủ tục đó phải được xây dựng, lập thành văn bản, thực hiện và duy trì. CHÚ THÍCH 2: Mức độ văn bản hóa hệ thống quản lý chất lượng của mỗi tổ chức có thể khác nhau tuỳ thuộc vào a) quy mô của tổ chức và loại hình hoạt động, b) sự phức tạp và sự tương tác giữa các quá trình, và c) năng lực của con người. CHÚ THÍCH 3: Hệ thống tài liệu có thể ở bất kỳ dạng hoặc loại phương tiện truyền thông nào. | 4.2. Yêu cầu về hệ thống tài liệu 4.2.1. Khái quát Các tài liệu của hệ thống quản lý chất lượng phải bao gồm: a) văn bản công bố về chính sách chất lượng và mục tiêu chất lượng; b) sổ tay chất lượng; c) các thủ tục dạng văn bản theo yêu cầu của tiêu chuẩn này; d) các tài liệu cần có của tổ chức để đảm bảo việc hoạch định, tác nghiệp và kiểm soát có hiệu lực các quá trình của tổ chức đó; e) các hồ sơ theo yêu cầu của tiêu chuẩn này (xem 4.2.4). f) mọi tài liệu khác được quy định bởi các văn bản pháp quy quốc gia hoặc khu vực. Ở trong tiêu chuẩn này, nếu có quy định rằng một yêu cầu, thủ tục, hoạt động hoặc thỏa thuận đặc biệt được "lập thành văn bản" thì yêu cầu, thủ tục, hoạt động hoặc thỏa thuận đặc biệt đó phải được áp dụng và duy trì. Đối với mỗi loại hoặc kiểu dụng cụ y tế, tổ chức phải thiết lập và duy trì một tệp tài liệu gồm có hoặc xác định các tài liệu định rõ quy định đối với sản phẩm và yêu cầu đối với hệ thống quản lý chất lượng (xem 4.2.3). Các tài liệu này định rõ quá trình chế tạo hoàn chỉnh và, nếu thích hợp, lắp đặt và dịch vụ bảo dưỡng. CHÚ THÍCH 1: Mức độ văn bản hóa hệ thống quản lý chất lượng của mỗi tổ chức có thể khác nhau tuỳ thuộc vào: a) quy mô của tổ chức và loại hình hoạt động; b) sự phức tạp và sự liên hệ giữa các quá trình; c) năng lực của con người. CHÚ THÍCH 2: Hệ thống tài liệu được thể hiện dưới bất kỳ dạng hoặc loại phương tiện truyền thông nào. Lý do về sự khác biệt: Nội dung của 4.2.1 của TCVN ISO 13485 bao gồm tất cả các yêu cầu có trong 4.2.1 của TCVN ISO 9001 tương ứng, có bổ sung thêm công bố chung liên quan đến những văn bản pháp quy mà có thể các yêu cầu về hệ thống tài liệu và yêu cầu đặc thù đối với tệp dữ liệu (file) chứa những tài liệu quy định cho từng loại/kiểu dụng cụ y tế. Ngoài ra, nội dung này còn bao gồm cả các yêu cầu về hệ thống tài liệu cho những hoạt động và bố trí đặc biệt. Nội dung này phù hợp hoàn toàn với các mục tiêu của việc thực hiện quy định của các văn bản pháp quy hiện hành và việc thúc đẩy sự hài hòa của những văn bản pháp quy mới liên quan đến dụng cụ y tế trên phạm vi toàn thế giới. | ||||||||
4.2.2. Sổ tay chất lượng Tổ chức phải lập và duy trì sổ tay chất lượng trong đó bao gồm a) phạm vi của hệ thống quản lý chất lượng, bao gồm cả các nội dung chi tiết và lý giải về bất cứ ngoại lệ nào (xem 1.2), b) các thủ tục dạng văn bản được thiết lập cho hệ thống quản lý chất lượng hoặc viện dẫn đến chúng và, c) mô tả sự tương tác giữa các quá trình trong hệ thống quản lý chất lượng. | 4.2.2. Sổ tay chất lượng Tổ chức phải lập và duy trì sổ tay chất lượng trong đó bao gồm: a) phạm vi của hệ thống quản lý chất lượng, bao gồm cả các nội dung chi tiết và sự giải thích về mọi ngoại lệ và/hoặc những quy định không áp dụng (xem 1.2); b) các thủ tục dạng văn bản được thiết lập cho hệ thống quản lý chất lượng hoặc viện dẫn đến chúng; c) mô tả sự tương tác giữa các quá trình trong hệ thống quản lý chất lượng. Sổ tay chất lượng cần nêu rõ cấu trúc của hệ thống tài liệu được sử dụng trong hệ thống quản lý chất lượng. | ||||||||
4.2.3. Kiểm soát tài liệu Các tài liệu theo yêu cầu của hệ thống quản lý chất lượng phải được kiểm soát. Hồ sơ chất lượng là một loại tàì liệu đặc biệt và phải được kiểm soát theo các yêu cầu nêu trong 4.2.4. Phải lập một thủ tục dạng văn bản để xác định việc kiểm soát cần thiết nhằm: a) phê duyệt tài liệu về sự thỏa đáng trước khi ban hành, b) xem xét, cập nhật khi cần và phê duyệt lại tài liệu, c) đảm bảo nhận biết được các thay đổi và tình trạng sửa đổi hiện hành của tài liệu, d) đảm bảo các bản của các tài liệu thích hợp sẵn có ở nơi sử dụng, e) đảm bảo tài liệu luôn rõ ràng, dễ nhận biết, f) đảm bảo các tài liệu có nguồn gốc bên ngoài được nhận biết và việc phân phối chúng được kiểm soát, và g) ngăn ngừa việc sử dụng vô tình các tài liệu lỗi thời và áp dụng các dấu hiệu nhận biết thích hợp nếu chúng được giữ lại vì mục đích nào đó. | 4.2.3. Kiểm soát tài liệu Các tài liệu theo yêu cầu của hệ thống quản lý chất lượng phải được kiểm soát. Hồ sơ chất lượng là một loại tàì liệu đặc biệt và phải được kiểm soát theo các yêu cầu nêu trong 4.2.4. Phải lập một thủ tục dạng văn bản để xác định việc kiểm soát cần thiết nhằm: a) xem xét và phê duyệt tài liệu về sự thỏa đáng trước khi ban hành; b) xem xét, cập nhật khi cần và phê duyệt lại tài liệu; c) đảm bảo nhận biết được các thay đổi và tình trạng sửa đổi hiện hành của tài liệu; d) đảm bảo các bản của các tài liệu thích hợp sẵn có ở nơi sử dụng; e) đảm bảo tài liệu luôn rõ ràng, dễ nhận biết; f) đảm bảo các tài liệu có nguồn gốc bên ngoài được nhận biết và việc phân phối chúng được kiểm soát; g) ngăn ngừa việc sử dụng vô tình các tài liệu lỗi thời và áp dụng các dấu hiệu nhận biết thích hợp nếu chúng được giữ lại vì mục đích nào đó. Tổ chức phải đảm bảo rằng những thay đổi trong tài liệu đều được xem xét và phê duyệt bởi những người đã phê duyệt tài liệu trước đó hoặc bởi những người được chỉ định khác có khả năng tiếp cận được với thông tin gốc thích hợp làm cơ sở cho việc đưa ra quyết định. Tổ chức phải xác định thời hạn lưu giữ đối với các tài liệu được kiểm soát đã lỗi thời, ít nhất một bản đối với từng tài liệu. Thời hạn này phải đảm bảo được rằng những tài liệu mà theo đó các dụng cụ y tế đã được chế tạo và thử nghiệm luôn sẵn có ít nhất trong thời gian sử dụng của dụng cụ y tế liên quan do tổ chức xác định nhưng không ngắn hơn thời hạn lưu giữ của bất kỳ hồ sơ nào (xem 4.2.4) hoặc thời hạn đã được quy định bởi yêu cầu chế định liên quan. | ||||||||
4.2.4. Kiểm soát hồ sơ Phải lập và duy trì các hồ sơ để cung cấp bằng chứng về sự phù hợp với các yêu cầu và hoạt động tác nghiệp có hiệu lực của hệ thống quản lý chất lượng. Các hồ sơ chất lượng phải rõ ràng, dễ nhận biết và dễ sử dụng. Phải lập một thủ tục bằng văn bản để xác định việc kiểm soát cần thiết đối với việc nhận biết, bảo quản, bảo vệ, sử dụng, xác định thời gian lưu giữ và hủy bỏ các hồ sơ chất lượng. | 4.2.4. Kiểm soát hồ sơ Phải lập và duy trì các hồ sơ để cung cấp bằng chứng về sự phù hợp với các yêu cầu và hoạt động tác nghiệp có hiệu lực của hệ thống quản lý chất lượng. Các hồ sơ chất lượng phải rõ ràng, dễ nhận biết và dễ sử dụng. Phải lập một thủ tục bằng văn bản để xác định việc kiểm soát cần thiết đối với việc nhận biết, bảo quản, bảo vệ, sử dụng, xác định thời gian lưu giữ và hủy bỏ các hồ sơ chất lượng. Tổ chức phải lưu giữ các hồ sơ trong thời hạn ít nhất là bằng thời hạn sử dụng của dụng cụ y tế mà tổ chức đã xác định nhưng không ngắn hơn thời hạn 2 năm kể từ ngày sản phẩm đó được đưa ra thị trường hoặc thời hạn đã được quy định bởi yêu cầu chế định liên quan. | ||||||||
5. Trách nhiệm của lãnh đạo 5.1. Cam kết của lãnh đạo Lãnh đạo cao nhất phải cung cấp bằng chứng về sự cam kết của mình đối với việc xây dựng và thực hiện hệ thống quản lý chất lượng và cải tiến thường xuyên hiệu lực của hệ thống đó bằng cách a) truyền đạt cho tổ chức về tầm quan trọng của việc đáp ứng khách hàng cũng như các yêu cầu của pháp luật và chế định, b) thiết lập chính sách chất lượng, c) đảm bảo việc thiết lập các mục tiêu chất lượng, d) tiến hành việc xem xét của lãnh đạo, và e) đảm bảo sẵn có các nguồn lực. | 5. Trách nhiệm của lãnh đạo 5.1. Cam kết của lãnh đạo Lãnh đạo cao nhất phải cung cấp bằng chứng về sự cam kết của mình đối với việc xây dựng và thực hiện hệ thống quản lý chất lượng và duy trì hiệu lực của hệ thống đó bằng cách: a) truyền đạt cho tổ chức về tầm quan trọng của việc đáp ứng khách hàng cũng như các yêu cầu của pháp luật và chế định; b) thiết lập chính sách chất lượng; c) đảm bảo việc thiết lập các mục tiêu chất lượng; d) tiến hành việc xem xét của lãnh đạo; e) đảm bảo sẵn có các nguồn lực. CHÚ THÍCH: Với mục đích của tiêu chuẩn này, các yêu cầu của pháp luật chỉ giới hạn ở độ an toàn và tính năng sử dụng của dụng cụ y tế. Lý do về sự khác biệt: Nội dung của điều này phù hợp hoàn toàn với mục tiêu của việc thực hiện quy định của văn bản pháp quy hiện hành và thúc đẩy sự hài hòa của các văn bản pháp quy mới liên quan đến dụng cụ y tế trên phạm vi toàn thế giới. Các văn bản pháp quy hiện hành hướng vào tính hiệu lực của hệ thống quản lý chất lượng nhằm chỉ sản xuất ra các sản phẩm an toàn và có hiệu lực. | ||||||||
5.2. Hướng vào khách hàng Lãnh đạo cao nhất phải đảm bảo rằng các yêu cầu của khách hàng được xác định và đáp ứng nhằm nâng cao sự thỏa mãn khách hàng (xem 7.2.1 và 8.2.1). | 5.2. Hướng vào khách hàng Lãnh đạo cao nhất phải đảm bảo rằng các yêu cầu của khách hàng được xác định và đáp ứng (xem 7.2.1 và 8.2.1). Lý do về sự khác biệt: Việc thể hiện nội dung điều này (so với TCVN ISO 9001) là phù hợp với quan quan điểm cho rằng sự thỏa mãn của khách hàng không phải là mục tiêu chế định thích hợp đối với dụng cụ y tế. Do đó, nội dung này phù hợp với mục tiêu của TCVN ISO 13485, đó là thúc đẩy sự hài hòa các văn bản pháp quy liên quan đến hệ thống quản lý chất lượng trên phạm vi toàn thế giới. | ||||||||
5.3. Chính sách chất lượng Lãnh đạo cao nhất phải đảm bảo rằng chính sách chất lượng a) phù hợp với mục đích của tổ chức, b) bao gồm việc cam kết đáp ứng các yêu cầu và cải tiến thường xuyên hiệu lực của hệ thống quản lý chất lượng, c) cung cấp cơ sở cho việc thiết lập và xem xét các mục tiêu chất lượng, d) được truyền đạt và thấu hiểu trong tổ chức, và e) được xem xét để luôn thích hợp. | 5.3. Chính sách chất lượng Lãnh đạo cao nhất phải đảm bảo rằng chính sách chất lượng: a) phù hợp với mục đích của tổ chức; b) bao gồm việc cam kết đáp ứng các yêu cầu và duy trì hiệu lực của hệ thống quản lý chất lượng; c) cung cấp cơ sở cho việc thiết lập và xem xét các mục tiêu chất lượng; d) được truyền đạt và thấu hiểu trong tổ chức; e) được xem xét để luôn thích hợp. Lý do về sự khác biệt: Nội dung 5.3 của TCVN ISO 13485 loại bỏ khỏi mục b) điều cam kết về cải tiến liên tục tính hiệu lực của hệ thống quản lý chất lượng và thay thế bằng lời cam kết về duy trì tính hiệu lực của hệ thống quản lý chất lượng. Sự thay thế này là phù hợp với mục tiêu của các văn bản pháp quy hiện hành và nhằm thúc đẩy sự hài hòa của những văn bản pháp quy liên quan đến hệ thống quản lý chất lượng trên phạm vi toàn thế giới. | ||||||||
| 5.4. Hoạch định [Nội dung của 5.4 của TCVN ISO 13485 hoàn toàn tương đương với nội dung của 5.4 của TCVN ISO 9001] | ||||||||
5.5. Trách nhiệm, quyền hạn và trao đổi thông tin 5.5.1. Trách nhiệm và quyền hạn Lãnh đạo cao nhất phải đảm bảo các trách nhiệm, quyền hạn và mối quan hệ của chúng được xác định và thông báo trong tổ chức. | 5.5. Trách nhiệm, quyền hạn và trao đổi thông tin 5.5.1. Trách nhiệm và quyền hạn Lãnh đạo cao nhất phải đảm bảo các trách nhiệm, quyền hạn và mối quan hệ của chúng được xác định, được lập thành văn bản và thông báo trong tổ chức. Lãnh đạo cao nhất phải thiết lập mối quan hệ tương tác giữa tất cả mọi người chịu sự lãnh đạo của mình, thực hiện và kiểm tra xác nhận công việc ảnh hưởng tới chất lượng và phải đảm bảo sự độc lập và quyền hạn cần thiết cho việc thực hiện những nhiệm vụ này. CHÚ THÍCH: Các văn bản pháp quy quốc gia hoặc khu vực có thể yêu cầu việc chỉ định những người cụ thể chịu trách nhiệm về các hoạt động liên quan đến việc theo dõi những sự kiện diễn ra ở giai đoạn tiền sản xuất và thông báo về những sự việc bất lợi (xem 8.2.1 và 8.5.1). | ||||||||
5.5.2. Đại diện của lãnh đạo Lãnh đạo cao nhất phải chỉ định một thành viên trong ban lãnh đạo, ngoài các trách nhiệm khác, có trách nhiệm và quyền hạn bao gồm a) đảm bảo các quá trình cần thiết của hệ thống quản lý chất lượng được thiết lập, thực hiện và duy trì; b) báo cáo cho lãnh đạo cao nhất về kết quả hoạt động của hệ thống quản lý chất lượng và về mọi nhu cầu cải tiến, và c) đảm bảo thúc đẩy toàn bộ tổ chức nhận thức được các yêu cầu của khách hàng. CHÚ THÍCH: Trách nhiệm của đại diện lãnh đạo về chất lượng có thể bao gồm cả quan hệ với bên ngoài về các vấn đề có liên quan đến hệ thống quản lý chất lượng. | 5.5.2. Đại diện của lãnh đạo Lãnh đạo cao nhất phải chỉ định một thành viên trong ban lãnh đạo, ngoài các trách nhiệm khác, có trách nhiệm và quyền hạn bao gồm a) đảm bảo các quá trình cần thiết của hệ thống quản lý chất lượng được thiết lập, thực hiện và duy trì; b) báo cáo cho lãnh đạo cao nhất về kết quả hoạt động của hệ thống quản lý chất lượng và về mọi nhu cầu cải tiến, và c) đảm bảo thúc đẩy toàn bộ tổ chức nhận thức được các yêu cầu chế định và các yêu cầu của khách hàng. CHÚ THÍCH: Trách nhiệm của đại diện lãnh đạo về chất lượng có thể bao gồm cả quan hệ với bên ngoài về các vấn đề có liên quan đến hệ thống quản lý chất lượng. | ||||||||
| 5.5.3. Trao đổi thông tin nội bộ [Nội dung của 5.5.3 của TCVN ISO 13485 hoàn toàn tương đương với nội dung của 5.5.3 của TCVN ISO 9001] | ||||||||
5.6. Xem xét của lãnh đạo | 5.6. Xem xét của lãnh đạo 5.6.1. Khái quát [Nội dung của 5.6.1 của TCVN ISO 13485 hoàn toàn tương đương với nội dung của 5.6.1 của TCVN ISO 9001] | ||||||||
5.6.2. Đầu vào của việc xem xét Đầu vào của việc xem xét của lãnh đạo phải bao gồm thông tin về a) kết quả của các cuộc đánh giá, b) phản hồi của khách hàng, c) việc thực hiện các quá trình và sự phù hợp của sản phẩm, d) tình trạng của các hành động khắc phục và phòng ngừa, e) các hành động tiếp theo từ các cuộc xem xét của lãnh đạo lần trước, f) những thay đổi có thể ảnh hưởng đến hệ thống quản lý chất lượng, và g) các khuyến nghị về cải tiến. | 5.6.2. Đầu vào của việc xem xét Đầu vào của việc xem xét của lãnh đạo phải bao gồm thông tin về a) kết quả của các cuộc đánh giá, b) phản hồi của khách hàng, c) việc thực hiện các quá trình và sự phù hợp của sản phẩm, d) tình trạng của các hành động khắc phục và phòng ngừa, e) các hành động tiếp theo từ các cuộc xem xét của lãnh đạo lần trước, f) những thay đổi có thể ảnh hưởng đến hệ thống quản lý chất lượng, và g) các khuyến nghị về cải tiến, h) các yêu cầu chế định mới hoặc đã được sửa đổi. | ||||||||
5.6.3. Đầu ra của việc xem xét Đầu ra của việc xem xét của lãnh đạo phải bao gồm mọi quyết định và hành động liên quan đến a) việc nâng cao tính hiệu lực của hệ thống quản lý chất lượng và cải tiến các quá trình của hệ thống, b) việc cải tiến các sản phẩm liên quan đến yêu cầu của khách hàng, và c) nhu cầu về nguồn lực. | 5.6.3. Đầu ra của việc xem xét Đầu ra của việc xem xét của lãnh đạo phải bao gồm mọi quyết định và hành động liên quan đến: a) các cải tiến cần thiết để duy trì tính hiệu lực của hệ thống quản lý chất lượng và các quá trình của hệ thống; b) việc cải tiến các sản phẩm liên quan đến yêu cầu của khách hàng; c) nhu cầu về nguồn lực. | ||||||||
6. Quản lý nguồn lực 6.1. Cung cấp nguồn lực Tổ chức phải xác định và cung cấp các nguồn lực cần thiết để a) thực hiện và duy trì hệ thống quản lý chất lượng và thường xuyên nâng cao hiệu lực của hệ thống đó, và b) tăng sự thỏa mãn khách hàng bằng cách đáp ứng các yêu cầu của khách hàng. | 6. Quản lý nguồn lực 6.1. Cung cấp nguồn lực Tổ chức phải xác định và cung cấp các nguồn lực cần thiết để: a) thực hiện và duy trì hệ thống quản lý chất lượng và duy trì hiệu lực của hệ thống đó; b) đáp ứng các yêu cầu chế định và yêu cầu của khách hàng. | ||||||||
6.2. Nguồn nhân lực | 6.2. Nguồn nhân lực 6.2.1. Khái quát [Nội dung của 6.2.1 của TCVN ISO 13485 hoàn toàn tương đương với nội dung của 6.2.1 của TCVN ISO 9001] | ||||||||
6.2.2. Năng lực, nhận thức và đào tạo Tổ chức phải a) xác định năng lực cần thiết của những người thực hiện các công việc ảnh hưởng đến chất lượng sản phẩm, b) tiến hành đào tạo hay những hành động khác để đáp ứng các nhu cầu này, c) đánh giá hiệu lực của các hành động được thực hiện, d) đảm bảo rằng người lao động nhận thức được mối liên quan và tầm quan trọng của các hoạt động của họ và họ đóng góp như thế nào đối với việc đạt được mục tiêu chất lượng, và e) duy trì hồ sơ thích hợp về giáo dục, đào tạo, kỹ năng và kinh nghiệm chuyên môn (xem 4.2.4). | 6.2.2. Năng lực, nhận thức và đào tạo Tổ chức phải a) xác định năng lực cần thiết của những người thực hiện các công việc ảnh hưởng đến chất lượng sản phẩm, b) tiến hành đào tạo hay những hành động khác để đáp ứng các nhu cầu này, c) đánh giá hiệu lực của các hành động được thực hiện, d) đảm bảo rằng người lao động nhận thức được mối liên quan và tầm quan trọng của các hoạt động của họ và họ đóng góp như thế nào đối với việc đạt được mục tiêu chất lượng, và e) duy trì hồ sơ thích hợp về giáo dục, đào tạo, kỹ năng và kinh nghiệm chuyên môn (xem 4.2.4). CHÚ THÍCH: Các văn bản pháp quy quốc gia hoặc khu vực có thể đòi hỏi tổ chức phải thiết lập những thủ tục dạng văn bản để xác định các nhu cầu đào tạo. | ||||||||
6.3. Cơ sở hạ tầng Tổ chức phải xác định, cung cấp và duy trì cơ sở hạ tầng cần thiết để đạt được sự phù hợp đối với các yêu cầu về sản phẩm. Cơ sở hạ tầng bao gồm ví dụ như: a) nhà cửa, không gian làm việc và các phương tiện kèm theo; b) trang thiết bị (cả phần cứng và phần mềm) và c) dịch vụ hỗ trợ (như vận chuyển hoặc trao đổi thông tin). | 6.3. Cơ sở hạ tầng Tổ chức phải xác định, cung cấp và duy trì cơ sở hạ tầng cần thiết để đạt được sự phù hợp đối với các yêu cầu về sản phẩm. Cơ sở hạ tầng bao gồm ví dụ như: a) nhà cửa, không gian làm việc và các phương tiện kèm theo; b) trang thiết bị (cả phần cứng và phần mềm) và c) dịch vụ hỗ trợ (như vận chuyển hoặc trao đổi thông tin). Tổ chức phải thiết lập các yêu cầu dạng văn bản đối với các hoạt động bảo dưỡng, kể cả tần suất thực hiện, khi những hoạt động đó hoặc khi việc không có chúng sẽ ảnh hưởng đến chất lượng sản phẩm. Hồ sơ của hoạt động bảo dưỡng phải được lưu giữ (xem 4.2.4). | ||||||||
6.4. Môi trường làm việc Tổ chức phải xác định và quản lý môi trường làm việc cần thiết để đạt được sự phù hợp đối với các yêu cầu của sản phẩm. | 6.4. Môi trường làm việc Tổ chức phải xác định và quản lý môi trường làm việc cần thiết để đạt được sự phù hợp đối với các yêu cầu của sản phẩm. Các yêu cầu sau phải áp dụng. a) Tổ chức phải thiết lập các yêu cầu dạng văn bản về sức khỏe, tình trạng sạch sẽ và quần áo của người lao động nếu sự tiếp xúc giữa họ với sản phẩm hoặc môi trường làm việc có thể có ảnh hưởng xấu tới chất lượng của sản phẩm (xem 7.5.1.2.1). b) Nếu điều kiện của môi trường làm việc có thể có tác động xấu đối với chất lượng sản phẩm, tổ chức phải thiết lập các yêu cầu dạng văn bản đối với điều kiện của môi trường làm việc đó và các thủ tục dạng văn bản hoặc hướng dẫn công việc để theo dõi và kiểm soát điều kiện của môi trường làm việc (xem 7.5.1.2.1). c) Tổ chức phải đảm bảo rằng mọi người lao động được yêu cầu làm việc tạm thời trong điều kiện môi trường đặc biệt của môi trường làm việc đều được đào tạo một cách phù hợp hoặc được giám sát bởi những người đã được đào tạo [xem 6.2.2 b)]. d) Nếu thích hợp, phải thiết lập và lập thành văn bản các thỏa thuận đặc biệt về kiểm soát sản phẩm bị nhiễm bẩn và có khả năng bị nhiễm bẩn nhằm ngăn ngừa sự nhiễm bẩn của sản phẩm khác, của môi trường làm việc hoặc người lao động (xem 7.5.3.1). | ||||||||
7. Tạo sản phẩm 7.1. Hoạch định việc tạo sản phẩm Tổ chức phải lập kế hoạch và triển khai các quá trình cần thiết đối với việc tạo sản phẩm. Hoạch định việc tạo sản phẩm phải nhất quán với các yêu cầu của các quá trình khác của hệ thống quản lý chất lượng (xem 4.1). Trong quá trình hoạch định việc tạo sản phẩm, khi thích hợp tổ chức phải xác định những điều sau đây: a) các mục tiêu chất lượng và các yêu cầu đối với sản phẩm; b) nhu cầu thiết lập các quá trình, tài liệu và việc cung cấp các nguồn lực cụ thể đối với sản phẩm; c) các hoạt động kiểm tra xác nhận, xác nhận giá trị sử dụng, các hoạt động theo dõi, kiểm tra và thử nghiệm cụ thể cần thiết đối với sản phẩm và các chuẩn mực chấp nhận sản phẩm; d) các hồ sơ cần thiết để cung cấp bằng chứng rằng các quá trình thực hiện và sản phẩm tạo thành đáp ứng các yêu cầu (xem 4.2.4). Đầu ra của việc hoạch định phải được thể hiện phù hợp với phương pháp tác nghiệp của tổ chức. CHÚ THÍCH 1: Tài liệu qui định các quá trình của hệ thống quản lý chất lượng (bao gồm cả các quá trình tạo sản phẩm) và các nguồn lực được sử dụng đối với một sản phẩm, dự án hay hợp đồng cụ thể có thể được coi như một kế hoạch chất lượng. CHÚ THÍCH 2: Tổ chức phải áp dụng các yêu cầu nêu trong 7.3 để triển khai quá trình tạo sản phẩm. | 7. Tạo sản phẩm 7.1. Hoạch định việc tạo sản phẩm Tổ chức phải lập kế hoạch và triển khai các quá trình cần thiết đối với việc tạo sản phẩm. Hoạch định việc tạo sản phẩm phải nhất quán với các yêu cầu của các quá trình khác của hệ thống quản lý chất lượng (xem 4.1). Trong quá trình hoạch định việc tạo sản phẩm, khi thích hợp tổ chức phải xác định những điều sau đây: a) các mục tiêu chất lượng và các yêu cầu đối với sản phẩm; b) nhu cầu thiết lập các quá trình, tài liệu và việc cung cấp các nguồn lực cụ thể đối với sản phẩm; c) các hoạt động kiểm tra xác nhận, xác nhận giá trị sử dụng, các hoạt động theo dõi, kiểm tra và thử nghiệm cụ thể cần thiết đối với sản phẩm và các chuẩn mực chấp nhận sản phẩm; d) các hồ sơ cần thiết để cung cấp bằng chứng rằng các quá trình thực hiện và sản phẩm tạo thành đáp ứng các yêu cầu (xem 4.2.4). Đầu ra của việc hoạch định phải được thể hiện phù hợp với phương pháp tác nghiệp của tổ chức. Tổ chức phải thiết lập các yêu cầu dạng văn bản đối với việc quản lý các rủi ro trong toàn bộ quá trình tạo sản phẩm. Hồ sơ liên quan đến quản lý rủi ro phải được lưu giữ (xem 4.2.4). CHÚ THÍCH 1: Tài liệu qui định các quá trình của hệ thống quản lý chất lượng (bao gồm cả các quá trình tạo sản phẩm) và các nguồn lực được sử dụng đối với một sản phẩm, dự án hay hợp đồng cụ thể có thể được coi như một kế hoạch chất lượng. CHÚ THÍCH 2: Tổ chức phải áp dụng các yêu cầu nêu trong 7.3 để triển khai quá trình tạo sản phẩm. CHÚ THÍCH 3: Xem ISO 14971 về hướng dẫn liên quan đến quản lý rủi ro. Lý do về sự khác biệt: Nhằm làm cho nội dung này phù hợp hoàn toàn với mục tiêu của việc thực hiện quy định của các văn bản pháp quy hiện hành và việc thúc đẩy sự hài hòa của các văn bản pháp quy mới liên quan đến dụng cụ y tế trên phạm vi toàn thế giới. Quản lý rủi ro là một hoạt động chủ yếu xác định bản chất và khối lượng công việc trong nhiều lĩnh vực mà hệ thống quản lý chất lượng của tổ chức đề cập đến. | ||||||||
7.2. Các quá trình liên quan đến khách hàng | 7.2. Các quá trình liên quan đến khách hàng 7.2.1. Xác định các yêu cầu liên quan đến sản phẩm [Nội dung của 7.2.1 của TCVN ISO 13485 hoàn toàn tương đương với nội dung của 7.2.1 của TCVN ISO 9001] | ||||||||
7.2.2. Xem xét các yêu cầu liên quan đến sản phẩm Tổ chức phải xem xét các yêu cầu liên quan đến sản phẩm. Việc xem xét này phải được tiến hành trước khi tổ chức cam kết cung cấp sản phẩm cho khách hàng (ví dụ như nộp đơn dự thầu, chấp nhận hợp đồng hay đơn đặt hàng, chấp nhận sự thay đổi trong hợp đồng hay đơn đặt hàng) và phải đảm bảo rằng a) yêu cầu về sản phẩm được định rõ; b) các yêu cầu trong hợp đồng hoặc đơn đặt hàng khác với những gì đã nêu trước đó phải được giải quyết; và c) tổ chức có khả năng đáp ứng các yêu cầu đã định. Phải duy trì hồ sơ các kết quả của việc xem xét và các hành động nảy sinh từ việc xem xét (xem 4.2.4). Khi khách hàng đưa ra các yêu cầu không bằng văn bản, các yêu cầu của khách hàng phải được tổ chức đó khẳng định trước khi chấp nhận. Khi yêu cầu về sản phẩm thay đổi, tổ chức phải đảm bảo rằng các văn bản tương ứng được sửa đổi và các cá nhân liên quan nhận thức được các yêu cầu thay đổi đó. CHÚ THÍCH: Trong một số tình huống, ví dụ như trong bán hàng qua internet, với mỗi lần đặt hàng, việc xem xét một cách chính thức là không thực tế. Thay vào đó, việc xem xét có thể được thực hiện đối với các thông tin thích hợp về sản phẩm như danh mục chào hàng hay tài liệu quảng cáo. | 7.2.2. Xem xét các yêu cầu liên quan đến sản phẩm Tổ chức phải xem xét các yêu cầu liên quan đến sản phẩm. Việc xem xét này phải được tiến hành trước khi tổ chức cam kết cung cấp sản phẩm cho khách hàng (ví dụ như nộp đơn dự thầu, chấp nhận hợp đồng hay đơn đặt hàng, chấp nhận sự thay đổi trong hợp đồng hay đơn đặt hàng) và phải đảm bảo rằng a) yêu cầu về sản phẩm được định rõ và được lập thành văn bản; b) các yêu cầu trong hợp đồng hoặc đơn đặt hàng khác với những gì đã nêu trước đó phải được giải quyết; và c) tổ chức có khả năng đáp ứng các yêu cầu đã định. Phải duy trì hồ sơ các kết quả của việc xem xét và các hành động nảy sinh từ việc xem xét (xem 4.2.4). Khi khách hàng đưa ra các yêu cầu không bằng văn bản, các yêu cầu của khách hàng phải được tổ chức đó khẳng định trước khi chấp nhận. Khi yêu cầu về sản phẩm thay đổi, tổ chức phải đảm bảo rằng các văn bản tương ứng được sửa đổi và các cá nhân liên quan nhận thức được các yêu cầu thay đổi đó. CHÚ THÍCH: Trong một số tình huống, ví dụ như trong bán hàng qua internet, với mỗi lần đặt hàng, việc xem xét một cách chính thức là không thực tế. Thay vào đó, việc xem xét có thể được thực hiện đối với các thông tin thích hợp về sản phẩm như danh mục chào hàng hay tài liệu quảng cáo. | ||||||||
7.2.3. Trao đổi thông tin với khách hàng Tổ chức phải xác định và sắp xếp có hiệu quả việc trao đổi thông tin với khách hàng có liên quan tới a) thông tin về sản phẩm; b) xử lý các yêu cầu, hợp đồng hoặc đơn đặt hàng, kể cả các sửa đổi, và c) phản hồi của khách hàng, kể cả các khiếu nại. | 7.2.3. Trao đổi thông tin với khách hàng Tổ chức phải xác định và sắp xếp có hiệu quả việc trao đổi thông tin với khách hàng có liên quan tới a) thông tin về sản phẩm; b) xử lý các yêu cầu, hợp đồng hoặc đơn đặt hàng, kể cả các sửa đổi; c) phản hồi của khách hàng, kể cả các khiếu nại; d) các thông báo tư vấn (xem 8.5.1). | ||||||||
7.3. Thiết kế và phát triển 7.3.1. Hoạch định thiết kế và phát triển Tổ chức phải lập kế hoạch và kiểm soát việc thiết kế và phát triển sản phẩm. Trong quá trình hoạch định thiết kế và phát triển tổ chức phải xác định a) các giai đoạn của thiết kế và phát triển, b) việc xem xét, kiểm tra xác nhận và xác nhận giá trị sử dụng thích hợp cho mỗi giai đoạn thiết kế và phát triển, và c) trách nhiệm và quyền hạn đối với các hoạt động thiết kế và phát triển. Tổ chức phải quản lý sự tương giao giữa các nhóm khác nhau tham dự vào việc thiết kế và phát triển nhằm đảm bảo sự trao đổi thông tin có hiệu quả và phân công trách nhiệm rõ ràng. Kết quả hoạch định phải được cập nhật một cách thích hợp trong quá trình thiết kế và phát triển. | 7.3. Thiết kế và phát triển 7.3.1. Hoạch định thiết kế và phát triển Tổ chức phải thiết lập các thủ tục dạng văn bản đối với thiết kế và phát triển. Tổ chức phải lập kế hoạch và kiểm soát việc thiết kế và phát triển sản phẩm. Trong quá trình hoạch định thiết kế và phát triển tổ chức phải xác định a) các giai đoạn của thiết kế và phát triển, b) các hoạt động xem xét, kiểm tra xác nhận, xác nhận giá trị sử dụng và chuyển giao thiết kế (xem CHÚ THÍCH) thích hợp cho mỗi giai đoạn thiết kế và phát triển, và c) trách nhiệm và quyền hạn đối với các hoạt động thiết kế và phát triển. Tổ chức phải quản lý sự tương giao giữa các nhóm khác nhau tham dự vào việc thiết kế và phát triển nhằm đảm bảo sự trao đổi thông tin có hiệu quả và phân công trách nhiệm rõ ràng. Hoạch định đầu ra phải được lập thành văn bản và được cập nhật theo tiến triển của hoạt động thiết kế và phát triển (xem 4.2.3). CHÚ THÍCH: Hoạt động chuyển giao thiết kế trong quá trình thiết kế và phát triển đảm bảo rằng đầu ra của thiết kế và phát triển được kiểm tra xác nhận là thích hợp với chế tạo sau khi trở thành quy định sản xuất cuối cùng. Lý do về sự khác biệt: Nội dung này phù hợp hoàn toàn với mục tiêu của việc thực hiện quy định của các văn bản pháp quy hiện hành và việc thúc đẩy sự hài hòa của các văn bản pháp quy mới liên quan đến dụng cụ y tế trên phạm vi toàn thế giới. Nhìn chung, TCVN ISO 13485 đưa ra mức độ yêu cầu đối với các thủ tục bằng văn bản tương tự như mức độ yêu cầu của TCVN ISO 9001:1994 - tiêu chuẩn phù hợp với nhiều văn bản pháp quy của nơi áp dụng. | ||||||||
7.3.2. Đầu vào của thiết kế và phát triển Đầu vào liên quan đến các yêu cầu đối với sản phẩm phải được xác định và duy trì hồ sơ (xem 4.2.4). Đầu vào phải bao gồm a) yêu cầu về chức năng và công dụng, b) yêu cầu chế định và luật pháp thích hợp, c) thông tin có thể áp dụng nhận được từ các thiết kế tương tự trước đó, và d) các yêu cầu khác cốt yếu cho thiết kế và phát triển. Những đầu vào này phải được xem xét về sự thích đáng. Những yêu cầu này phải đầy đủ, không mơ hồ và không mâu thuẫn với nhau. | 7.3.2. Đầu vào của thiết kế và phát triển Đầu vào liên quan đến các yêu cầu đối với sản phẩm phải được xác định và duy trì hồ sơ (xem 4.2.4). Đầu vào phải bao gồm a) yêu cầu về chức năng, hoạt động và an toàn theo công dụng, b) yêu cầu chế định và luật pháp thích hợp, c) thông tin có thể áp dụng nhận được từ các thiết kế tương tự trước đó, và d) các yêu cầu khác cốt yếu cho thiết kế và phát triển, e) đầu ra của quản lý rủi ro (xem 7.1). Đầu vào này phải được xem xét về sự phù hợp và phải được chấp thuận. Những đầu vào này phải được xem xét về sự thích đáng. Những yêu cầu này phải đầy đủ, không mơ hồ và không mâu thuẫn với nhau. | ||||||||
7.3.3. Đầu ra của thiết kế và phát triển Đầu ra của thiết kế và phát triển phải ở dạng sao cho có thể kiểm tra xác nhận theo đầu vào của thiết kế và phát triển và phải được phê duyệt trước khi ban hành. Đầu ra của thiết kế và phát triển phải a) đáp ứng các yêu cầu đầu vào của thiết kế và phát triển, b) cung cấp các thông tin thích hợp cho việc mua hàng, sản xuất và cung cấp dịch vụ, c) bao gồm hoặc viện dẫn tới các chuẩn mực chấp nhận của sản phẩm, và d) xác định các đặc tính cốt yếu cho an toàn và sử dụng đúng của sản phẩm. | 7.3.3. Đầu ra của thiết kế và phát triển Đầu ra của thiết kế và phát triển phải ở dạng sao cho có thể kiểm tra xác nhận theo đầu vào của thiết kế và phát triển và phải được phê duyệt trước khi ban hành. Đầu ra của thiết kế và phát triển phải: a) đáp ứng các yêu cầu đầu vào của thiết kế và phát triển; b) cung cấp các thông tin thích hợp cho việc mua hàng, sản xuất và cung cấp dịch vụ; c) bao gồm hoặc viện dẫn tới các chuẩn mực chấp nhận của sản phẩm; d) xác định các đặc tính cốt yếu cho an toàn và sử dụng đúng của sản phẩm. Các hồ sơ về đầu ra của thiết kế và phát triển phải được lưu giữ (xem 4.2.4). CHÚ THÍCH: Các hồ sơ về đầu ra của thiết kế và phát triển có thể bao gồm quy định kỹ thuật, quy trình chế tạo, bản vẽ kỹ thuật và nhật ký kỹ thuật hoặc nghiên cứu. | ||||||||
7.3.4. Xem xét thiết kế và phát triển Tại những giai đoạn thích hợp, việc xem xét thiết kế và phát triển một cách có hệ thống phải được thực hiện theo hoạch định để a) đánh giá khả năng đáp ứng các yêu cầu của các kết quả thiết kế và phát triển, và b) nhận biết mọi vấn đề trục trặc và đề xuất các hành động cần thiết. Những người tham dự vào việc xem xét phải bao gồm đại diện của tất cả các bộ phận chức năng liên quan tới các giai đoạn thiết kế và phát triển đang được xem xét. Phải duy trì hồ sơ về các kết quả xem xét và mọi hành động cần thiết (xem 4.2.4). | 7.3.4. Xem xét thiết kế và phát triển Tại những giai đoạn thích hợp, việc xem xét thiết kế và phát triển một cách có hệ thống phải được thực hiện theo hoạch định để a) đánh giá khả năng đáp ứng các yêu cầu của các kết quả thiết kế và phát triển, và b) nhận biết mọi vấn đề trục trặc và đề xuất các hành động cần thiết. Những người tham dự vào việc xem xét phải bao gồm đại diện của tất cả các bộ phận chức năng liên quan tới các giai đoạn thiết kế và phát triển đang được xem xét cũng như các chuyên gia khác.(xem 5.5.1 và 6.2.1). Phải duy trì hồ sơ về các kết quả xem xét và mọi hành động cần thiết (xem 4.2.4). | ||||||||
| 7.3.5. Kiểm tra xác nhận thiết kế và phát triển [Nội dung của 7.3.5 của TCVN ISO 13485 hoàn toàn tương đương với nội dung của 7.3.5 của TCVN ISO 9001] | ||||||||
7.3.6. Xác nhận giá trị sử dụng của thiết kế và phát triển Xác nhận giá trị sử dụng của thiết kế và phát triển phải được tiến hành theo các bố trí đã hoạch định (xem 7.3.1) để đảm bảo rằng sản phẩm tạo ra có khả năng đáp ứng các yêu cầu sử dụng dự kiến hay các ứng dụng qui định khi đã biết. Khi có thể, phải tiến hành xác nhận giá trị sử dụng trước khi chuyển giao hay sử dụng sản phẩm. Phải duy trì hồ sơ các kết quả của việc xác nhận giá trị sử dụng và mọi hành động cần thiết (xem 4.2.4). | 7.3.6. Xác nhận giá trị sử dụng của thiết kế và phát triển Xác nhận giá trị sử dụng của thiết kế và phát triển phải được tiến hành theo các bố trí đã hoạch định (xem 7.3.1) để đảm bảo rằng sản phẩm tạo ra có khả năng đáp ứng các yêu cầu sử dụng dự kiến hay các ứng dụng qui định khi đã biết. Phải tiến hành xác nhận giá trị sử dụng trước khi chuyển giao hay sử dụng sản phẩm (xem CHÚ THÍCH 1). Phải duy trì hồ sơ các kết quả của việc xác nhận giá trị sử dụng và mọi hành động cần thiết (xem 4.2.4). Tổ chức phải thực hiện các đánh giá về khả năng sử dụng và/hoặc đánh giá về tính năng của dụng cụ y tế như là một hoạt động thành phần của việc xác nhận giá trị sử dụng của thiết kế và phát triển, theo yêu cầu của các văn bản pháp quy quốc gia hoặc khu vực (xem CHÚ THÍCH 2). CHÚ THÍCH 1: Nếu dụng cụ y tế chỉ có thể được xác nhận giá trị sử dụng sau khi lắp ráp và lắp đặt tại nơi sử dụng, việc chuyển giao chưa được coi là hoàn thành cho tới khi sản phẩm này đã chính thức được giao cho khách hàng. CHÚ THÍCH 2: Việc cung cấp dụng cụ y tế cho mục đích đánh giá khả năng sử dụng và/hoặc đánh giá tính năng không được coi là hoạt động chuyển giao sản phẩm. | ||||||||
| 7.3.7. Kiểm soát thay đổi thiết kế và phát triển [Nội dung của 7.3.7 của TCVN ISO 13485 hoàn toàn tương đương với nội dung của 7.3.7 của TCVN ISO 9001] | ||||||||
7.4. Mua hàng 7.4.1. Quá trình mua hàng Tổ chức phải đảm bảo sản phẩm mua vào phù hợp với các yêu cầu mua sản phẩm đã qui định. Cách thức và mức độ kiểm soát áp dụng cho người cung ứng và sản phẩm mua vào phụ thuộc vào sự tác động của sản phẩm mua vào đối với việc tạo ra sản phẩm tiếp theo hay thành phẩm. Tổ chức phải đánh giá và lựa chọn người cung ứng dựa trên khả năng cung cấp sản phẩm phù hợp với các yêu cầu của tổ chức. Phải xác định các chuẩn mực lựa chọn, đánh giá và đánh giá lại. Phải duy trì hồ sơ các kết quả của việc đánh giá và mọi hành động cần thiết nảy sinh từ việc đánh giá (xem 4.2.4). | 7.4. Mua hàng 7.4.1. Quá trình mua hàng Tổ chức phải thiết lập các thủ tục dạng văn bản để đảm bảo rằng sản phẩm mua vào phù hợp với các yêu cầu mua sản phẩm đã qui định. Cách thức và mức độ kiểm soát áp dụng cho người cung ứng và sản phẩm mua vào phụ thuộc vào sự tác động của sản phẩm mua vào đối với việc tạo ra sản phẩm tiếp theo hay thành phẩm. Tổ chức phải đánh giá và lựa chọn người cung ứng dựa trên khả năng cung cấp sản phẩm phù hợp với các yêu cầu của tổ chức. Phải xác định các chuẩn mực lựa chọn, đánh giá và đánh giá lại. Phải duy trì hồ sơ các kết quả của việc đánh giá và mọi hành động cần thiết nảy sinh từ việc đánh giá (xem 4.2.4). | ||||||||
7.4.2. Thông tin mua hàng Thông tin mua hàng phải miêu tả sản phẩm được mua, nếu thích hợp có thể bao gồm a) yêu cầu về phê duyệt sản phẩm, các thủ tục, quá trình, và thiết bị, b) yêu cầu về trình độ con người, và c) yêu cầu về hệ thống quản lý chất lượng. Tổ chức phải đảm bảo sự thỏa đáng của các yêu cầu mua hàng đã qui định trước khi thông báo cho người cung ứng. | 7.4.2. Thông tin mua hàng Thông tin mua hàng phải miêu tả sản phẩm được mua, nếu thích hợp có thể bao gồm: a) yêu cầu về phê duyệt sản phẩm, các thủ tục, quá trình, và thiết bị; b) yêu cầu về trình độ con người; c) yêu cầu về hệ thống quản lý chất lượng. Tổ chức phải đảm bảo sự thỏa đáng của các yêu cầu mua hàng đã qui định trước khi thông báo cho người cung ứng. Theo mức độ yêu cầu về xác định nguồn gốc nêu trong 7.5.3.2, tổ chức phải duy trì các thông tin mua hàng liên quan, nghĩa là các tài liệu (xem 4.2.3) và hồ sơ (xem 4.2.4). | ||||||||
7.4.3. Kiểm tra xác nhận sản phẩm mua vào Tổ chức phải lập và thực hiện các hoạt động kiểm tra hoặc các hoạt động khác cần thiết để đảm bảo rằng sản phẩm mua vào đáp ứng các yêu cầu mua hàng đã qui định. Khi tổ chức hoặc khách hàng có ý định thực hiện các hoạt động kiểm tra xác nhận tại cơ sở của người cung ứng, tổ chức phải công bố việc sắp xếp kiểm tra xác nhận dự kiến và phương pháp thông qua sản phẩm trong các thông tin mua hàng. | 7.4.3. Kiểm tra xác nhận sản phẩm mua vào Tổ chức phải lập và thực hiện các hoạt động kiểm tra hoặc các hoạt động khác cần thiết để đảm bảo rằng sản phẩm mua vào đáp ứng các yêu cầu mua hàng đã qui định. Khi tổ chức hoặc khách hàng có ý định thực hiện các hoạt động kiểm tra xác nhận tại cơ sở của người cung ứng, tổ chức phải công bố việc sắp xếp kiểm tra xác nhận dự kiến và phương pháp thông qua sản phẩm trong các thông tin mua hàng. Các hồ sơ của kiểm tra xác nhận phải được lưu giữ (xem 4.2.4). | ||||||||
7.5. Sản xuất và cung cấp dịch vụ 7.5.1. Kiểm soát sản xuất và cung cấp dịch vụ Tổ chức phải lập kế hoạch, tiến hành sản xuất và cung cấp dịch vụ trong điều kiện được kiểm soát. Khi có thể, các điều kiện được kiểm soát phải bao gồm a) sự sẵn có các thông tin mô tả các đặc tính của sản phẩm, b) sự sẵn có các hướng dẫn công việc khi cần, c) việc sử dụng các thiết bị thích hợp, d) sự sẵn có và việc sử dụng các phương tiện theo dõi và đo lường, e) thực hiện việc đo lường và theo dõi, và f) thực hiện các hoạt động thông qua, giao hàng và các hoạt động sau giao hàng. | 7.5. Sản xuất và cung cấp dịch vụ 7.5.1. Kiểm soát sản xuất và cung cấp dịch vụ 7.5.1.1. Yêu cầu chung Tổ chức phải lập kế hoạch, tiến hành sản xuất và cung cấp dịch vụ trong điều kiện được kiểm soát. Khi có thể, các điều kiện được kiểm soát phải bao gồm: a) sự sẵn có các thông tin mô tả các đặc tính của sản phẩm, b) sự sẵn có các hướng dẫn công việc khi cần, c) việc sử dụng các thiết bị thích hợp, d) sự sẵn có và việc sử dụng các phương tiện theo dõi và đo lường, e) thực hiện việc đo lường và theo dõi, f) thực hiện các hoạt động thông qua, giao hàng và các hoạt động sau giao hàng; g) thực hiện các thao tác đã xác định về ghi nhãn và bao gói. Tổ chức phải thiết lập và duy trì hồ sơ (xem 4.2.4) đối với từng mẻ dụng cụ y tế để xác định nguồn gốc ở mức độ quy định ở 7.5.3 và xác định lượng sản phẩm chế tạo và lượng sản phẩm chấp thuận phân phối. Hồ sơ về mẻ sản phẩm phải được kiểm tra xác nhận và phê duyệt. CHÚ THÍCH: Một mẻ có thể là một dụng cụ y tế đơn lẻ. 7.5.1.2. Kiểm soát sản xuất và cung cấp dịch vụ - Yêu cầu riêng 7.5.1.2.1. Độ sạch của sản phẩm và kiểm soát sự nhiễm bẩn Tổ chức phải thiết lập các yêu cầu dạng văn bản đối với độ sạch của sản phẩm nếu a) sản phẩm được tổ chức làm sạch trước khi khử trùng và/hoặc sử dụng, hoặc b) sản phẩm được cung ứng ở trạng thái chưa khử trùng và sẽ được đưa vào quá trình làm sạch trước khi khử trùng và/hoặc sử dụng, hoặc c) sản phẩm được cung ứng để sử dụng ở trạng thái chưa khử trùng và độ sạch của sản phẩm là rất quan trọng trong sử dụng, hoặc d) các tác nhân của quá trình được tách khỏi sản phẩm trong quá trình chế tạo. Nếu sản phẩm được làm sạch theo a) hoặc b) nêu ở trên, các yêu cầu ở 6.4 a) và 6.4 b) không áp dụng trước cho quá trình làm sạch. 7.5.1.2.2. Hoạt động lắp đặt Nếu phù hợp, tổ chức phải thiết lập các yêu cầu dạng văn bản trong đó có những chuẩn cứ chấp nhận đối với việc lắp đặt và kiểm tra xác nhận việc lắp đặt dụng cụ y tế. Nếu yêu cầu đã được khách hàng đồng ý cho phép tổ chức khác hoặc đơn vị được tổ chức đó ủy quyền, thực hiện việc lắp đặt, tổ chức phải cung cấp các yêu cầu dạng văn bản đối với lắp đặt và kiểm tra xác nhận. Hồ sơ về lắp đặt và kiểm tra xác nhận do tổ chức hoặc đơn vị được tổ chức ủy quyền thực hiện phải được lưu giữ (xem 4.2.4). 7.5.1.2.3. Hoạt động dịch vụ Nếu việc cung cấp dịch vụ là yêu cầu quy định thì tổ chức phải thiết lập các thủ tục dạng văn bản, hướng dẫn công việc, tài liệu tham chiếu và quy trình đo chuẩn cần thiết để thực hiện hoạt động dịch vụ và kiểm tra xác nhận sự đáp ứng yêu cầu quy định của hoạt động dịch vụ. Hồ sơ về hoạt động dịch vụ do tổ chức thực hiện phải được lưu giữ (xem 4,2,4). CHÚ THÍCH: Ví dụ, dịch vụ có thể bao gồm sửa chữa và bảo dưỡng. 7.5.1.3. Yêu cầu cụ thể đối với dụng cụ y tế vô trùng Tổ chức phải duy trì hồ sơ về các tham số của quá trình đối với quá trình khử trùng đã được sử dụng cho từng mẻ khử trùng (xem 4.2.4). Hồ sơ về khử trùng phải có khả năng xác định nguồn gốc đối với từng mẻ sản xuất dụng cụ y tế (xem 7.5.1.1). | ||||||||
7.5.2. Xác nhận giá trị sử dụng của các quá trình sản xuất và cung cấp dịch vụ Tổ chức phải xác nhận giá trị sử dụng đối với của mọi quá trình sản xuất và cung cấp dịch vụ có kết quả đầu ra không thể kiểm tra xác nhận bằng cách theo dõi hoặc đo lường sau đó. Điều này bao gồm mọi quá trình mà sự sai sót chỉ có thể trở nên rõ ràng sau khi sản phẩm được sử dụng hoặc dịch vụ được chuyển giao. Việc xác nhận giá trị sử dụng phải chứng tỏ khả năng của các quá trình để đạt được kết quả đã hoạch định. Đối với các quá trình đó, khi có thể, tổ chức phải sắp xếp những điều sau: a) các chuẩn mực đã định để xem xét và phê duyệt các quá trình, b) phê duyệt thiết bị và trình độ con người, c) sử dụng các phương pháp và thủ tục cụ thể, d) các yêu cầu về hồ sơ (xem 4.2.4); và e) tái xác nhận giá trị sử dụng. | 7.5.2. Xác nhận giá trị sử dụng của các quá trình sản xuất và cung cấp dịch vụ 7.5.2.1. Yêu cầu chung Tổ chức phải xác nhận giá trị sử dụng đối với của mọi quá trình sản xuất và cung cấp dịch vụ có kết quả đầu ra không thể kiểm tra xác nhận bằng cách theo dõi hoặc đo lường sau đó. Điều này bao gồm mọi quá trình mà sự sai sót chỉ có thể trở nên rõ ràng sau khi sản phẩm được sử dụng hoặc dịch vụ được chuyển giao. Việc xác nhận giá trị sử dụng phải chứng tỏ khả năng của các quá trình để đạt được kết quả đã hoạch định. Đối với các quá trình đó, khi có thể, tổ chức phải sắp xếp những điều sau: a) các chuẩn mực đã định để xem xét và phê duyệt các quá trình; b) phê duyệt thiết bị và trình độ con người; c) sử dụng các phương pháp và thủ tục cụ thể; d) các yêu cầu về hồ sơ (xem 4.2.4); e) tái xác nhận giá trị sử dụng. Tổ chức phải thiết lập các thủ tục dạng văn bản đối với việc xác nhận giá trị sử dụng của ứng dụng phần mềm máy tính (và những thay đổi đối với phần mềm đó và/hoặc ứng dụng của nó) đối với việc sản xuất và cung cấp dịch vụ mà có ảnh hưởng tới khả năng phù hợp với yêu cầu quy định của sản phẩm. ứng dụng phần mềm đó phải được xác nhận giá trị sử dụng trước khi sử dụng lần đầu. Hồ sơ về xác nhận giá trị sử dụng phải được lưu giữ (xem 4.2.4). 7.5.2.2. Yêu cầu cụ thể đối với dụng cụ y tế vô trùng Tổ chức phải thiết lập các thủ tục dạng văn bản đối với việc xác nhận giá trị sử dụng của các quá trình khử trùng. Quá trình khử trùng phải được xác nhận giá trị sử dụng trước khi sử dụng lần đầu. Hồ sơ về xác nhận giá trị sử dụng của mỗi quá trình khử trùng phải được lưu giữ (xem 4.2.4). | ||||||||
7.5.3. Nhận biết và xác định nguồn gốc Khi cần thiết, tổ chức phải nhận biết sản phẩm bằng các biện pháp thích hợp trong suốt quá trình tạo sản phẩm. Tổ chức phải nhận biết được trạng thái của sản phẩm tương ứng với các yêu cầu theo dõi và đo lường. Tổ chức phải kiểm soát và lưu hồ sơ việc nhận biết duy nhất sản phẩm khi việc xác định nguồn gốc là một yêu cầu (xem 4.2.4). CHÚ THÍCH: Trong một số lĩnh vực công nghiệp, quản lý cấu hình là phương pháp để duy trì việc nhận biết và xác định nguồn gốc. | 7.5.3. Nhận biết và xác định nguồn gốc 7.5.3.1. Nhận biết Tổ chức phải nhận biết sản phẩm bằng các biện pháp thích hợp trong suốt quá trình tạo sản phẩm và phải thiết lập thủ tục dạng văn bản cho việc nhận biết sản phẩm. Tổ chức phải thiết lập các thủ tục dạng văn bản để đảm bảo rằng dụng cụ y tế được hoàn trả lại tổ chức sẽ được nhận biết và phân biệt được với sản phẩm phù hợp [xem 6.4 d)]. 7.5.3.2. Xác định nguồn gốc 7.5.3.2.1. Quy định chung Tổ chức phải thiết lập các thủ tục dạng văn bản đối với việc xác định nguồn gốc. Những thủ tục đó phải xác định mức độ xác định nguồn gốc của sản phẩm và các hồ sơ yêu cầu (xem 4.2.4, 8.3 và 8.5). Tổ chức phải kiểm soát và lưu hồ sơ việc nhận biết duy nhất sản phẩm khi việc xác định nguồn gốc là một yêu cầu (xem 4.2.4). CHÚ THÍCH: Quản lý cấu hình là phương pháp để duy trì việc nhận biết và xác định nguồn gốc. 7.5.3.2.2. Yêu cầu cụ thể đối với dụng cụ y tế cấy ghép hoạt tính và dụng cụ y tế cấy ghép Để xác định các hồ sơ yêu cầu về xác định nguồn gốc, tổ chức phải có hồ sơ về tất cả các bộ phận cấu thành, vật liệu và điều kiện của môi trường làm việc nếu chúng là nguyên nhân làm cho dụng cụ y tế không thỏa mãn yêu cầu quy định. Tổ chức phải yêu cầu các đại lý hoặc các nhà phân phối của mình duy trì hồ sơ về việc phân phối dụng cụ y tế để cho phép xác định nguồn gốc và những hồ sơ đó phải luôn sẵn có để phục vụ cho việc kiểm tra. Hồ sơ về tên gọi và địa chỉ của bên nhận hàng phải được lưu giữ (xem 4.2.4). 7.5.3.3. Nhận biết trạng thái Tổ chức phải nhận biết được trạng thái của sản phẩm tương ứng với các yêu cầu theo dõi và đo lường. Việc nhận biết trạng thái của sản phẩm phải được duy trì trong suốt quá trình sản xuất, bảo quản, lắp đặt và cung cấp dịch vụ để đảm bảo rằng chỉ có sản phẩm đã qua kiểm tra và thử nghiệm (hoặc được nghiệm thu trong điều kiện nhượng bộ được phép) mới được gửi đi, sử dụng hoặc lắp đặt. | ||||||||
7.5.4. Tài sản của khách hàng Tổ chức phải gìn giữ tài sản của khách hàng khi chúng thuộc sự kiểm soát của tổ chức hay được tổ chức sử dụng. Tổ chức phải nhận biết, kiểm tra xác nhận, bảo vệ tài sản do khách hàng cung cấp để sử dụng hoặc để hợp thành sản phẩm. Bất kỳ tài sản nào của khách hàng bị mất mát, hư hỏng hoặc được phát hiện không phù hợp cho việc sử dụng đều phải được thông báo cho khách hàng và các hồ sơ phải được duy trì (xem 4.2.4). CHÚ THÍCH: Tài sản của khách hàng có thể bao gồm cả sở hữu trí tuệ. | 7.5.4. Tài sản của khách hàng Tổ chức phải gìn giữ tài sản của khách hàng khi chúng thuộc sự kiểm soát của tổ chức hay được tổ chức sử dụng. Tổ chức phải nhận biết, kiểm tra xác nhận, bảo vệ tài sản do khách hàng cung cấp để sử dụng hoặc để hợp thành sản phẩm. Bất kỳ tài sản nào của khách hàng bị mất mát, hư hỏng hoặc được phát hiện không phù hợp cho việc sử dụng đều phải được thông báo cho khách hàng và các hồ sơ phải được duy trì (xem 4.2.4). CHÚ THÍCH: Tài sản của khách hàng có thể bao gồm cả sở hữu trí tuệ. | ||||||||
7.5.5. Bảo toàn sản phẩm Tổ chức phải bảo toàn sự phù hợp của sản phẩm trong suốt các quá trình nội bộ và giao hàng đến vị trí đã định. Việc bảo toàn này phải bao gồm nhận biết, xếp dỡ (di chuyển), bao gói, lưu giữ, bảo quản. Việc bảo toàn cũng phải áp dụng với các bộ phận cấu thành của sản phẩm. | 7.5.5. Bảo toàn sản phẩm Tổ chức phải thiết lập các thủ tục dạng văn bản hoặc hướng dẫn công việc dạng văn bản về bảo toàn sự phù hợp của sản phẩm trong suốt các quá trình nội bộ và giao hàng đến vị trí đã định. Việc bảo toàn này phải bao gồm nhận biết, xếp dỡ (di chuyển), bao gói, lưu giữ, bảo quản. Việc bảo toàn cũng phải áp dụng với các bộ phận cấu thành của sản phẩm. Tổ chức phải thiết lập các thủ tục dạng văn bản hoặc hướng dẫn công việc dạng văn bản về kiểm soát sản phẩm theo thời hạn sử dụng giới hạn hoặc điều kiện bảo quản đặc biệt yêu cầu. Điều kiện bảo quản đặc biệt đó phải được kiểm soát và lập thành hồ sơ (xem 4.2.4). | ||||||||
7.6. Kiểm soát phương tiện theo dõi và đo lường Tổ chức phải xác định việc theo dõi và đo lường cần thực hiện và các phương tiện theo dõi và đo lường cần thiết để cung cấp bằng chứng về sự phù hợp của sản phẩm với các yêu cầu đã xác định (xem 7.2.1). Tổ chức phải thiết lập các quá trình để đảm bảo rằng việc theo dõi và đo lường có thể tiến hành và được tiến hành một cách nhất quán với các yêu cầu theo dõi và đo lường. Khi cần thiết để đảm bảo kết quả đúng, thiết bị đo lường phải a) được hiệu chuẩn hoặc kiểm tra xác nhận định kỳ, hoặc trước khi sử dụng, dựa trên các chuẩn đo lường có liên kết được với chuẩn đo lường quốc gia hay quốc tế; khi không có các chuẩn này thì căn cứ được sử dụng để hiệu chuẩn hoặc kiểm tra xác nhận phải được lưu hồ sơ; b) được hiệu chỉnh hoặc hiệu chỉnh lại, khi cần thiết; c) được nhận biết để giúp xác định trạng thái hiệu chuẩn; d) được giữ gìn tránh bị hiệu chỉnh làm mất tính đúng đắn của các kết quả đo; e) được bảo vệ để tránh hư hỏng hoặc suy giảm chất lượng trong khi di chuyển, bảo dưỡng và lưu giữ. Ngoài ra, tổ chức phải đánh giá và ghi nhận giá trị hiệu lực của các kết quả đo lường trước đó khi thiết bị được phát hiện không phù hợp với yêu cầu. Tổ chức phải tiến hành các hành động thích hợp đối với thiết bị đó và bất kỳ sản phẩm nào bị ảnh hưởng. Phải duy trì hồ sơ (xem 4.2.4) của kết quả hiệu chuẩn và kiểm tra xác nhận. Khi sử dụng phần mềm máy tính để theo dõi và đo lường các yêu cầu đã qui định, phải khẳng định khả năng thỏa mãn việc áp dụng nhằm tới của chúng. Việc này phải được tiến hành trước lần sử dụng đầu tiên và được xác nhận lại khi cần thiết. CHÚ THÍCH: Xem hướng dẫn trong ISO 10012-1 và ISO 10012-2. | 7.6. Kiểm soát phương tiện theo dõi và đo lường Tổ chức phải xác định việc theo dõi và đo lường cần thực hiện và các phương tiện theo dõi và đo lường cần thiết để cung cấp bằng chứng về sự phù hợp của sản phẩm với các yêu cầu đã xác định (xem 7.2.1). Tổ chức phải thiết lập các thủ tục dạng văn bản để đảm bảo rằng việc theo dõi và đo lường có thể tiến hành và được tiến hành một cách nhất quán với các yêu cầu theo dõi và đo lường. Khi cần thiết để đảm bảo kết quả đúng, thiết bị đo lường phải: a) được hiệu chuẩn hoặc kiểm tra xác nhận định kỳ, hoặc trước khi sử dụng, dựa trên các chuẩn đo lường có liên kết được với chuẩn đo lường quốc gia hay quốc tế; khi không có các chuẩn này thì căn cứ được sử dụng để hiệu chuẩn hoặc kiểm tra xác nhận phải được lưu hồ sơ; b) được hiệu chỉnh hoặc hiệu chỉnh lại, khi cần thiết; c) được nhận biết để giúp xác định trạng thái hiệu chuẩn; d) được giữ gìn tránh bị hiệu chỉnh làm mất tính đúng đắn của các kết quả đo; e) được bảo vệ để tránh hư hỏng hoặc suy giảm chất lượng trong khi di chuyển, bảo dưỡng và lưu giữ. Ngoài ra, tổ chức phải đánh giá và ghi nhận giá trị hiệu lực của các kết quả đo lường trước đó khi thiết bị được phát hiện không phù hợp với yêu cầu. Tổ chức phải tiến hành các hành động thích hợp đối với thiết bị đó và bất kỳ sản phẩm nào bị ảnh hưởng. Phải duy trì hồ sơ (xem 4.2.4) của kết quả hiệu chuẩn và kiểm tra xác nhận. Khi sử dụng phần mềm máy tính để theo dõi và đo lường các yêu cầu đã qui định, phải khẳng định khả năng thỏa mãn việc áp dụng nhằm tới của chúng. Việc này phải được tiến hành trước lần sử dụng đầu tiên và được xác nhận lại khi cần thiết. CHÚ THÍCH: Xem hướng dẫn trong ISO 10012 liên quan đến hệ thống quản lý đo lường. | ||||||||
8. Đo lường, phân tích và cải tiến 8.1. Khái quát Tổ chức phải hoạch định và triển khai các quá trình theo dõi, đo lường, phân tích và cải tiến cần thiết để a) chứng tỏ sự phù hợp của sản phẩm, b) đảm bảo sự phù hợp của hệ thống quản lý chất lượng, và c) thường xuyên nâng cao tính hiệu lực của hệ thống quản lý chất lượng. Điều này phải bao gồm việc xác định các phương pháp có thể áp dụng, kể cả các kỹ thuật thống kê, và mức độ sử dụng chúng. | 8. Đo lường, phân tích và cải tiến 8.1. Khái quát Tổ chức phải hoạch định và triển khai các quá trình theo dõi, đo lường, phân tích và cải tiến cần thiết để: a) chứng tỏ sự phù hợp của sản phẩm; b) đảm bảo sự phù hợp của hệ thống quản lý chất lượng; c) duy trì tính hiệu lực của hệ thống quản lý chất lượng. Điều này phải bao gồm việc xác định các phương pháp có thể áp dụng, kể cả các kỹ thuật thống kê, và mức độ sử dụng chúng. CHÚ THÍCH: Các văn bản pháp quy quốc giá hoặc khu vực có thể yêu cầu những thủ tục dạng văn bản về áp dụng và kiểm soát việc ứng dụng các kỹ thuật thống kê. Lý do về sự khác biệt: Nhằm làm cho nội dung này phù hợp hoàn toàn với mục tiêu của việc thực hiện quy định của các văn bản pháp quy hiện hành và việc thúc đẩy sự hài hòa của các văn bản pháp quy mới liên quan đến dụng cụ y tế trên phạm vi toàn thế giới. Mục tiêu của các văn bản pháp quy liên quan đến dụng cụ y tế là duy trì tính hiệu lực của hệ thống quản lý chất lượng nhằm chỉ sản xuất ra những sản phẩm an toàn và có hiệu lực chứ không phải là cải tiến liên tục hệ thống quản lý chất lượng. | ||||||||
8.2. Theo dõi và đo lường 8.2.1. Sự thỏa mãn của khách hàng Tổ chức phải theo dõi các thông tin về sự chấp nhận của khách hàng về việc tổ chức có đáp ứng yêu cầu của khách hàng hay không, coi đó như một trong những thước đo mức độ thực hiện của hệ thống quản lý chất lượng. Phải xác định các phương pháp để thu thập và sử dụng các thông tin này. | 8.2. Theo dõi và đo lường 8.2.1. Thông tin phản hồi Tổ chức phải theo dõi các thông tin liên quan đến việc tổ chức có đáp ứng yêu cầu của khách hàng hay không, coi đó như một trong những thước đo mức độ thực hiện của hệ thống quản lý chất lượng. Phải xác định các phương pháp để thu thập và sử dụng các thông tin này. Tổ chức phải thiết lập thủ tục dạng văn bản đối với hệ thống phản hồi thông tin [xem 7.2.3 c)] để đưa ra cảnh báo sớm về các vấn đề chất lượng và đối với đầu vào của các quá trình của hành động khắc phục và phòng ngừa (xem 8.5.2 và 8.5.3). Nếu các văn bản pháp quy quốc gia hoặc khu vực yêu cầu tổ chức phải tích luỹ kinh nghiệm của giai đoạn tiền sản xuất thì việc xem xét kinh nghiệm này phải là một phần của hệ thống phản hồi thông tin đó (xem 8.5.1). Lý do về sự khác biệt: Các yêu cầu về "sự thỏa mãn của khách hàng" và "sự chấp nhận của khách hàng" đều mang tính rất chủ quan đối với việc áp dụng giống như các yêu cầu của một văn bản pháp quy. Nội dung này phù hợp với mục tiêu của việc thực hiện quy định của các văn bản pháp quy hiện hành và việc thúc đẩy sự hài hòa của các văn bản pháp quy mới liên quan đến dụng cụ y tế trên phạm vi toàn thế giới. | ||||||||
| 8.2.2. Đánh giá nội bộ [Nội dung của 8.2.2 của TCVN ISO 13485 hoàn toàn tương đương với nội dung của 8.2.2 của TCVN ISO 9001] | ||||||||
8.2.4. Theo dõi và đo lường sản phẩm Tổ chức phải theo dõi và đo lường các đặc tính của sản phẩm để kiểm tra xác nhận rằng các yêu cầu về sản phẩm được đáp ứng. Việc này phải được tiến hành tại những giai đoạn thích hợp của quá trình tạo sản phẩm theo các sắp xếp hoạch định (xem 7.1). Bằng chứng của sự phù hợp với các chuẩn mực chấp nhận phải được duy trì. Hồ sơ phải chỉ ra người có quyền hạn trong việc thông qua sản phẩm (xem 4.2.4). Chỉ được thông qua sản phẩm và chuyển giao dịch vụ khi đã hoàn thành thỏa đáng các hoạt động theo hoạch định (xem 7.1), nếu không phải được sự phê duyệt của người có thẩm quyền và, nếu có thể, của khách hàng. | 8.2.4. Theo dõi và đo lường sản phẩm 8.2.4.1. Yêu cầu chung Tổ chức phải theo dõi và đo lường các đặc tính của sản phẩm để kiểm tra xác nhận rằng các yêu cầu về sản phẩm được đáp ứng. Việc này phải được tiến hành tại những giai đoạn thích hợp của quá trình tạo sản phẩm theo các sắp xếp hoạch định (xem 7.1) và các thủ tục dạng văn bản (xem 7.5.1.1). Bằng chứng của sự phù hợp với các chuẩn mực chấp nhận phải được duy trì. Hồ sơ phải chỉ ra người có quyền hạn trong việc thông qua sản phẩm (xem 4.2.4). Chỉ được thông qua sản phẩm và chuyển giao dịch vụ khi đã hoàn thành thỏa đáng các hoạt động theo hoạch định (xem 7.1). 8.2.4.2. Yêu cầu cụ thể đối với dụng cụ y tế cấy ghép hoạt tính và dụng cụ y tế cấy ghép Tổ chức phải lập hồ sơ (xem 4.2.4) nhân dạng của những người thực hiện các phép kiểm tra hoặc thử nghiệm. | ||||||||
8.3. Kiểm soát sản phẩm không phù hợp Tổ chức phải đảm bảo rằng sản phẩm không phù hợp với các yêu cầu được nhận biết và kiểm soát để phòng ngừa việc sử dụng hoặc chuyển giao vô tình. Phải xác định trong một thủ tục dạng văn bản việc kiểm soát, các trách nhiệm và quyền hạn có liên quan đối với sản phẩm không phù hợp. Tổ chức phải xử lý sản phẩm không phù hợp bằng một hoặc một số cách sau: a) tiến hành loại bỏ sự không phù hợp được phát hiện; b) cho phép sử dụng, thông qua hoặc chấp nhận có nhân nhượng bởi người có thẩm quyền và, khi có thể, bởi khách hàng; c) tiến hành loại bỏ khỏi việc sử dụng hoặc áp dụng dự kiến ban đầu. Phải duy trì hồ sơ (xem 4.2.4) về bản chất các sự không phù hợp và bất kỳ hành động tiếp theo nào được tiến hành, kể cả các nhân nhượng có được. Khi sản phẩm không phù hợp được khắc phục, chúng phải được kiểm tra xác nhận lại để chứng tỏ sự phù hợp với các yêu cầu. Khi sản phẩm không phù hợp được phát hiện sau khi chuyển giao hoặc đã bắt đầu sử dụng, tổ chức phải có các hành động thích hợp đối với các tác động hoặc hậu quả tiềm ẩn của sự không phù hợp. | 8.3. Kiểm soát sản phẩm không phù hợp Tổ chức phải đảm bảo rằng sản phẩm không phù hợp với các yêu cầu được nhận biết và kiểm soát để phòng ngừa việc sử dụng hoặc chuyển giao vô tình. Phải xác định trong một thủ tục dạng văn bản việc kiểm soát, các trách nhiệm và quyền hạn có liên quan đối với sản phẩm không phù hợp. Tổ chức phải xử lý sản phẩm không phù hợp bằng một hoặc một số cách sau: a) tiến hành loại bỏ sự không phù hợp được phát hiện; b) cho phép sử dụng, thông qua hoặc chấp nhận có nhân nhượng; c) tiến hành loại bỏ khỏi việc sử dụng hoặc áp dụng dự kiến ban đầu. Tổ chức phải đảm bảo rằng sản phẩm không phù hợp chỉ được chấp nhận do nhân nhượng nếu đáp ứng yêu cầu chế định. Hồ sơ nhân dạng của những người cho phép nhân nhượng phải được lưu giữ (xem 4.2.4). Phải duy trì hồ sơ (xem 4.2.4) về bản chất các sự không phù hợp và bất kỳ hành động tiếp theo nào được tiến hành, kể cả các nhân nhượng có được. Khi sản phẩm không phù hợp được khắc phục, chúng phải được kiểm tra xác nhận lại để chứng tỏ sự phù hợp với các yêu cầu. Khi sản phẩm không phù hợp được phát hiện sau khi chuyển giao hoặc đã bắt đầu sử dụng, tổ chức phải có các hành động thích hợp đối với các tác động hoặc hậu quả tiềm ẩn của sự không phù hợp. Nếu cần phải làm lại sản phẩm (một lần hoặc nhiều lần), tổ chức phải lập thành văn bản quá trình làm lại đó dưới hình thức hướng dẫn công việc và tài liệu này phải trải qua cùng một thủ tục cho phép và phê duyệt như đối với hướng dẫn công việc ban đầu. Trước khi cho phép và phê duyệt hướng dẫn công việc đó, việc xác định mọi ảnh hưởng xấu của quá trình làm lại đối với sản phẩm phải được thực hiện và lập thành văn bản (xem 4.2.3 và 7.5.1). | ||||||||
8.4. Phân tích dữ liệu Tổ chức phải xác định, thu thập và phân tích các dữ liệu tương ứng để chứng tỏ sự thích hợp và tính hiệu lực của hệ thống quản lý chất lượng và đánh giá xem sự cải tiến thường xuyên hiệu lực của hệ thống chất lượng có thể tiến hành ở đâu. Điều này bao gồm cả các dữ liệu được tạo ra do kết quả của việc theo dõi, đo lường và từ các nguồn thích hợp khác. Việc phân tích dữ liệu phải cung cấp thông tin về: a) sự thỏa mãn khách hàng (xem 8.2.1); b) sự phù hợp với các yêu cầu về sản phẩm (xem 7.2.1); c) đặc tính và xu hướng của các quá trình và sản phẩm, kể cả các cơ hội cho hành động phòng ngừa, và d) người cung ứng. | 8.4. Phân tích dữ liệu Tổ chức phải thiết lập các thủ tục dạng văn bản để xác định, thu thập và phân tích các dữ liệu tương ứng để chứng tỏ sự thích hợp và tính hiệu lực của hệ thống quản lý chất lượng và đánh giá xem sự cải tiến hiệu lực của hệ thống chất lượng có thể tiến hành hay không. Điều này bao gồm cả các dữ liệu được tạo ra do kết quả của việc theo dõi, đo lường và từ các nguồn thích hợp khác. Việc phân tích dữ liệu phải cung cấp thông tin về: a) thông tin phản hồi (xem 8.2.1); b) sự phù hợp với các yêu cầu về sản phẩm (xem 7.2.1); c) đặc tính và xu hướng của các quá trình và sản phẩm, kể cả các cơ hội cho hành động phòng ngừa; d) người cung ứng. Hồ sơ về kết quả phân tích dữ liệu phải được lưu giữ (xem 4.2.4). | ||||||||
8.5. Cải tiến 8.5.1. Cải tiến thường xuyên Tổ chức phải thường xuyên nâng cao tính hiệu lực của hệ thống quản lý chất lượng thông qua việc sử dụng chính sách chất lượng, mục tiêu chất lượng, kết quả đánh giá, việc phân tích dữ liệu, hành động khắc phục và phòng ngừa và sự xem xét của lãnh đạo. | 8.5. Cải tiến 8.5.1. Khái quát Tổ chức phải nhận biết và thực hiện mọi thay đổi cần thiết để đảm bảo và duy trì sự phù hợp liên tục và hiệu lực của hệ thống quản lý chất lượng thông qua việc sử dụng chính sách chất lượng, mục tiêu chất lượng, kết quả đánh giá, việc phân tích dữ liệu, hành động khắc phục và phòng ngừa và sự xem xét của lãnh đạo. Tổ chức phải thiết lập các thủ tục dạng văn bản đối với việc ban hành và áp dụng các thông báo tư vấn. Các thủ tục đó phải có khả năng áp dụng ở mọi lúc. Hồ sơ của tất cả các cuộc điều tra về khiếu nại của khách hàng đều phải được lưu giữ (xem 4.2.4). Nếu cuộc điều tra xác định rằng hoạt động bên ngoài tổ chức dẫn tới khiếu nại của khách hàng thì thông tin liên quan phải được trao đổi giữa các tổ chức liên quan (xem 4.1). Nếu sau bất kỳ khiếu nại nào của khách hàng mà không có hành động khắc phục và/hoặc phòng ngừa thì nguyên nhân của việc không thực hiện hành động này phải được xác nhận (xem 5.5.1) và lập thành hồ sơ (4.2.4). Nếu các văn bản pháp quy quốc gia hoặc khu vực yêu cầu phải thông báo về những sự việc có ảnh hưởng xấu đáp ứng chuẩn cứ thông báo quy định, tổ chức phải thiết lập các thủ tục dạng văn bản đối với việc thông báo đến các cơ quan quản lý có thẩm quyền. Lý do về sự khác biệt: Nhằm làm cho nội dung này phù hợp với mục tiêu của việc thực hiện quy định của các văn bản pháp quy hiện hành và việc thúc đẩy sự hài hòa của các văn bản pháp quy mới liên quan đến dụng cụ y tế trên phạm vi toàn thế giới. Cải tiến liên tục hệ thống quản lý chất lượng không phải là mục tiêu hiện thời của các văn bản pháp quy. | ||||||||
8.5.2. Hành động khắc phục Tổ chức phải thực hiện hành động nhằm loại bỏ nguyên nhân của sự không phù hợp để ngăn ngừa sự tái diễn. Hành động khắc phục phải tương ứng với tác động của sự không phù hợp gặp phải. Phải lập một thủ tục dạng văn bản để xác định các yêu cầu về a) việc xem xét sự không phù hợp (kể cả các khiếu nại của khách hàng), b) việc xác định nguyên nhân của sự không phù hợp, c) việc đánh giá cần có các hành động để đảm bảo rằng sự không phù hợp không tái diễn, d) việc xác định và thực hiện các hành động cần thiết, e) việc lưu hồ sơ các kết quả của hành động được thực hiện (xem 4.2.4), và f) việc xem xét các hành động khắc phục đã thực hiện. | 8.5.2. Hành động khắc phục Tổ chức phải thực hiện hành động nhằm loại bỏ nguyên nhân của sự không phù hợp để ngăn ngừa sự tái diễn. Hành động khắc phục phải tương ứng với tác động của sự không phù hợp gặp phải. Phải lập một thủ tục dạng văn bản để xác định các yêu cầu về: a) việc xem xét sự không phù hợp (kể cả các khiếu nại của khách hàng); b) việc xác định nguyên nhân của sự không phù hợp; c) việc đánh giá cần có các hành động để đảm bảo rằng sự không phù hợp không tái diễn; d) việc xác định và thực hiện các hành động cần thiết, bao gồm, nếu phù hợp, việc cập nhật hệ thống tài liệu; e) việc lưu hồ sơ các kết quả của mọi cuộc điều tra và hành động được thực hiện (xem 4.2.4); f) việc xem xét các hành động khắc phục đã thực hiện và hiệu lực của hành động này. | ||||||||
8.5.3. Hành động phòng ngừa Tổ chức phải xác định các hành động nhằm loại bỏ nguyên nhân của sự không phù hợp tiềm ẩn để ngăn chặn sự xuất hiện của chúng. Các hành động phòng ngừa được tiến hành phải tương ứng với tác động của các vấn đề tiềm ẩn. Phải lập một thủ tục dạng văn bản để xác định các yêu cầu đối với a) việc xác định sự không phù hợp tiềm ẩn và các nguyên nhân của chúng, b) việc đánh giá nhu cầu thực hiện các hành động để phòng ngừa việc xuất hiện sự không phù hợp, c) việc xác định và thực hiện các hành động cần thiết, d) hồ sơ các kết quả của hành động được thực hiện (xem 4.2.4), và e) việc xem xét các hành động phòng ngừa được thực hiện. | 8.5.3. Hành động phòng ngừa Tổ chức phải xác định các hành động nhằm loại bỏ nguyên nhân của sự không phù hợp tiềm ẩn để ngăn chặn sự xuất hiện của chúng. Các hành động phòng ngừa được tiến hành phải tương ứng với tác động của các vấn đề tiềm ẩn. Phải lập một thủ tục dạng văn bản để xác định các yêu cầu đối với: a) việc xác định sự không phù hợp tiềm ẩn và các nguyên nhân của chúng; b) việc đánh giá nhu cầu thực hiện các hành động để phòng ngừa việc xuất hiện sự không phù hợp; c) việc xác định và thực hiện các hành động cần thiết; d) việc lưu hồ sơ các kết quả của mọi cuộc điều tra và hành động được thực hiện (xem 4.2.4); e) việc xem xét các hành động phòng ngừa được thực hiện và hiệu lực của hành động này. |
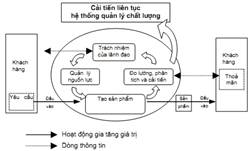
TÀI LIỆU THAM KHẢO
[1] TCVN ISO 9001:2000 (TCVN ISO 9001:2000), Hệ thống quản lý chất lượng - Các yêu cầu
[2] ISO 10012, Measurement management systems - Requirements for measurement processes and measuring equipment
[3] ISO 11134:1994, Sterilization of health care products - Requirements for validation and rountine control - Industrial moist heat sterilization
[4] ISO 11135:1994, Medical devices - Validation and rountine control of ethylene oxide sterilization (Corrigendum 1 published 1994)
[5] ISO 11137:1995, Sterilization of health care products - Requirements for validation and rountine control - Radiation sterilization (Corrigendum 1 published 1995; Amendment 1 published 2001)
[6] ISO 13641:2002, Elimination or reduction of risk of infection related to in vitro diagnostic medical devices
[7] ISO 13683:1997, Sterilization of health care products - Requirements for validation and rountine control of moist heat sterilization in health care facilities
[8] ISO 14155-1:2003, Clinical investigation of medical devices for human subjects - Part 1: General requirements
[9] ISO 14155-2:2003, Clinical investigation of medical devices for human subjects - Part 2: Clinical investigation plans
[10] ISO 14160:1998, Sterilization of medical devices - Validation and rountine control of sterilization of single-use medical devices incorporating materials of animal origin by liquid chemical sterilants
[11] ISO 14937:2000, Sterilization of health care products - General requirements for characterization of a sterilizing agent and the development, validation and rountine control of a sterilizing agent
[12] ISO/TR 14969, Medical devices - Quality management systems - Guidance on application of ISO 13485:2003
[13] ISO 14971:2000, Medical devices - Application of risk management to medical devices
[14] TCVN ISO 19011:2003 (ISO 19011:2002), Hướng dẫn đánh giá hệ thống quản lý chất lượng và/hoặc hệ thống quản lý môi trường
[15] Global Harmonization Task Force (GHTF) - Study Group 1 (SG 1), Document No29R11, dated 2 Feb.2002






